本页面已设置访问密码,请输入密码查看全文:
本文档结合了基因组复杂性评估标准以及多维数据综合判定方法。
基因组 Survey 的核心目的
基因组调研(Genome Survey)是基因组组装前的“勘探”阶段。其核心目标是评估基因组复杂程度,为后续的测序策略(如测序深度、库型选择)和组装算法提供科学依据。
- 高杂合度影响:导致姊妹染色体无法合并,组装结果倾向于大于实际基因组(冗余)。
- 高重复序列影响:导致组装过程中序列折叠或产生缺口(Gaps),组装结果倾向于小于实际基因组(碎片化)。
基因组复杂程度判定标准
基于测序数据,我们通过以下指标划分基因组的复杂等级:
经验性评价指标
|复杂度等级|杂合度 (ρ)|重复序列比例|GC 含量| |—|—|—|—| |简单基因组|$< 0.5\%$|$< 50\%$|$35\% - 65\%$| |复杂基因组|$0.5\% - 1.2\%$|$50\% - 65\%$|$< 35\%$ 或 $> 65\%$| |高度复杂基因组|$> 1.2\%$|$> 65\%$|极端 GC 分布|
注:多倍体基因组(如三倍体、四倍体)通常直接归类为复杂或高度复杂基因组。
杂合度 (Heterozygosity, ρ)
- 生物学定义:在二倍体或多倍体生物中,同源染色体上对应位点的等位基因(Allele)存在序列差异的程度。
- 数学表示:通常指随机选择两个同源位点时,碱基不同的概率。若杂合率为 1%,意味着平均每 100 个碱基中存在一个变异位点(如 SNP 或 Indel)。
- 对测序的影响:杂合位点会导致测序 Reads 被分为两群不同的序列,在 k-mer 分布中产生 1/2 频数的次峰。
重复序列 (Repetitive Sequences)
- 生物学定义:在单倍体基因组中,以多个拷贝形式存在的 DNA 序列。
- 分类:
- 散在重复序列:如转座子(TEs),随机分布在基因组各处。
- 串联重复序列:如微卫星 DNA(SSR),呈首尾相连状排列。
- 对测序的影响:由于序列高度相似,大量 Reads 会来源于这些拷贝,在 k-mer 分布中产生高频波峰(2M,3M,…)或明显的右侧拖尾。
K-mer 分析原理与数学基础
什么是 K-mer?
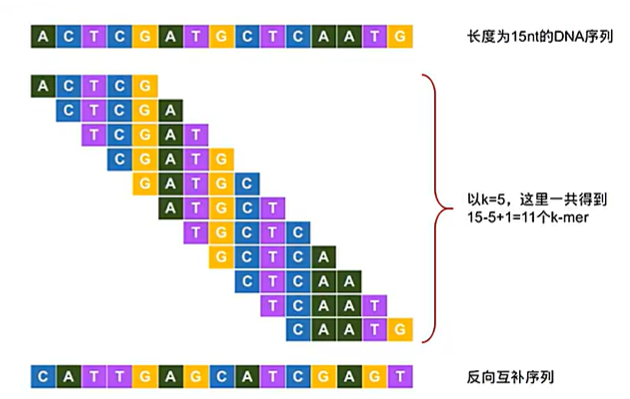
K-mer 是指从测序 Read 中截取的长度为 $K$ 的连续子串。
-
数量计算:一段长度为 $L$ 的 Read,可以产生的 K-mer 数量为 $L - K + 1$。
-
$K$ 取值原则:通常取奇数(如 17, 19, 21),目的是为了避免在处理反向互补序列时产生歧义。
-
多样性上限:对于长度为 $K$ 的 K-mer,理论上存在 $4^K$ 种排列组合。
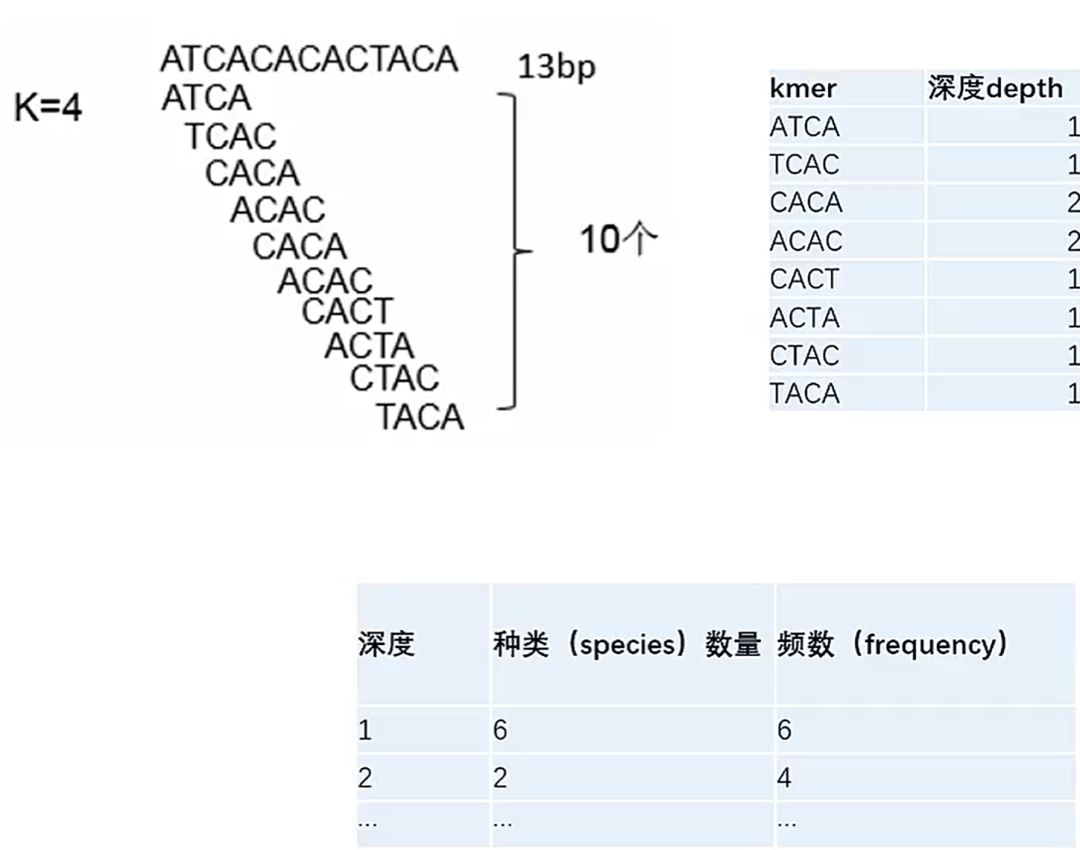
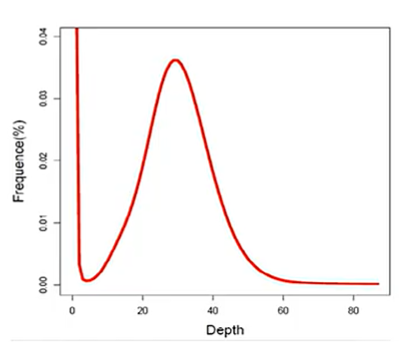
K-mer 频数分布图 (K-mer Species Profile)
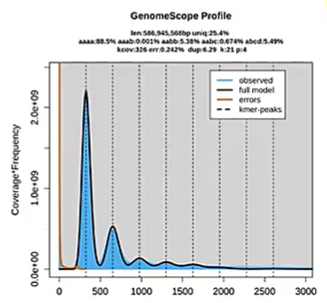
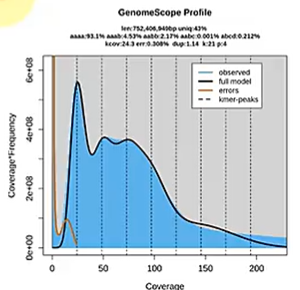
通过统计所有 K-mer 出现的频数(Depth),以频数为横坐标,该频数对应的 K-mer 种类数为纵坐标,绘制分布图。在理想状态(无错误、无重复、纯合)下,该图符合泊松分布。
波峰解读
在分析 K-mer 图时,我们需要针对观测到的不同波峰提出竞争性假说,并寻找诊断性证据。
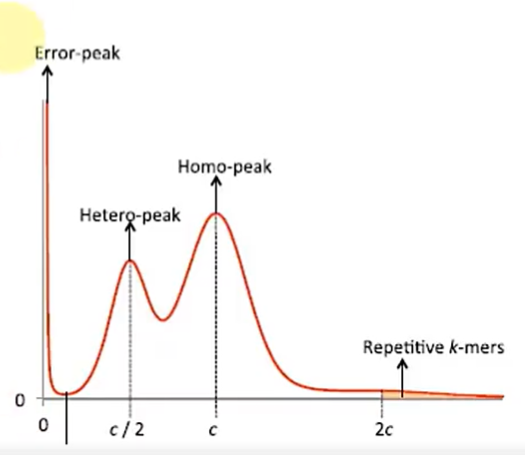
错误峰 (Error Peak)
- 位置:通常位于频数 $1 - 2$ 处。
- 原因:随机测序错误产生的 K-mer 通常只出现极少数次数。
- 处理:在进行数学拟合时需排除此区域。
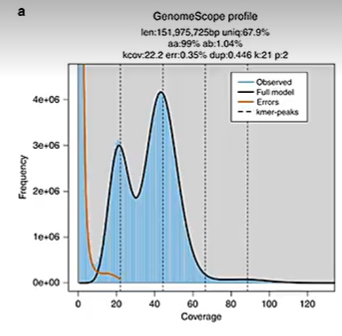
主峰 (Main/Homozygous Peak)
- 位置:位于平均测序深度 $M$ 处。
- 意义:代表基因组中单拷贝且纯合的序列。
杂合峰 (Heterozygous Peak)
- 位置:位于 $1/2 M$ 处。
- 诊断逻辑:若此处出现独立峰,说明大量 K-mer 的来源频率仅为总深度的一半,这是杂合性的直接证据。杂合度越高,该峰越高。
重复峰与拖尾 (Repetitive Peak & Tail)
- 位置:位于 $2M, 3M, \dots nM$ 处,或在主峰右侧形成长尾。
- 诊断逻辑:反映了在基因组中多次出现的序列。右侧曲线下面积(AUC)占比越大,重复率越高。
倍性判定:高级分布特征
不同倍性的基因组在 K-mer 分布图上具有独特的“指纹”:
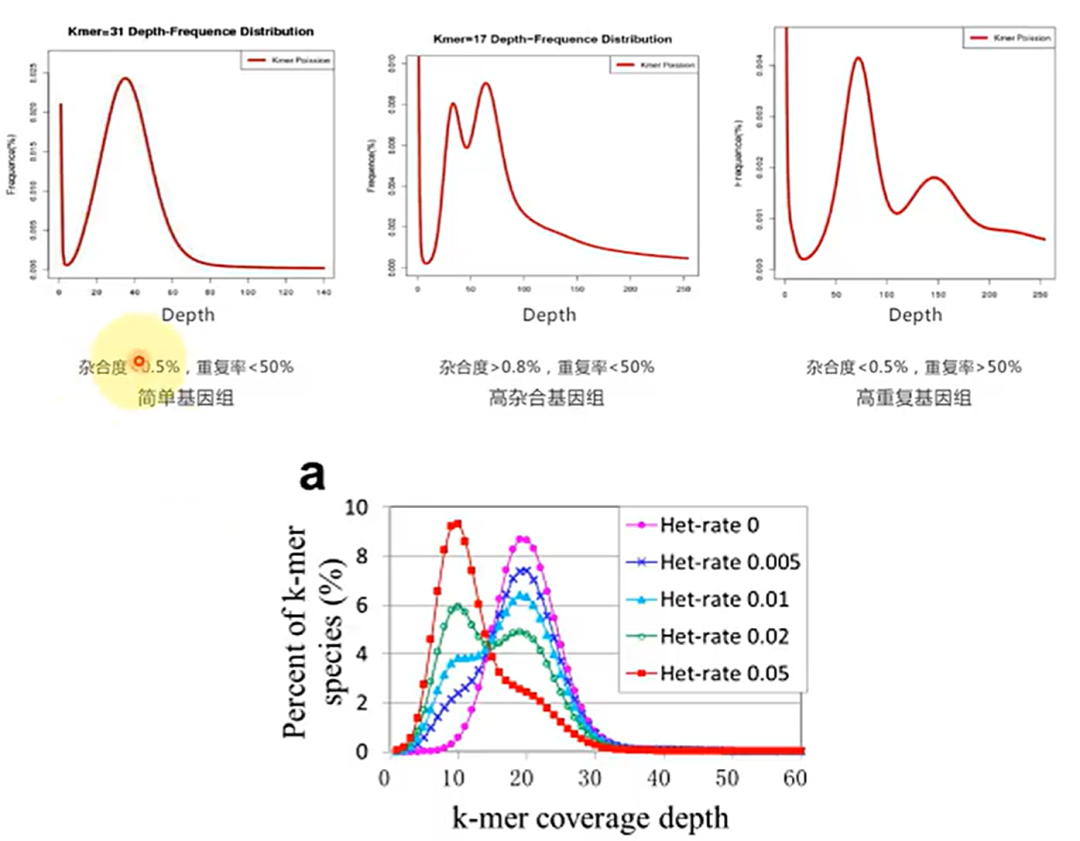
- 纯合二倍体:仅有一个清晰的主峰。
- 杂合二倍体:在 $1/2 M$ 和 $M$ 处有两个峰。
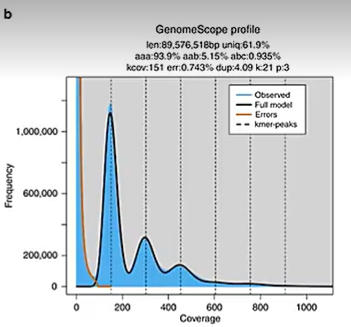
- 杂合三倍体:可能在 $1/3 M, 2/3 M, M$ 处出现多个峰值,峰高比例反映了等位基因的分布。
-
异源多倍体:由于不同基因组间的差异,波峰分布会更加复杂,通常表现为多个重叠的泊松分布。
- 异源杂合四倍体
1 | |
比对报告中看到高 Duplication Rate 时
假说 A:技术冗余(PCR Duplicates)
- 来源:文库构建过程中的 PCR 过度扩增。
- 逻辑:样本序列中存在大量物理意义上完全一致的分子。
- 诊断特征:1)K-mer 图的主峰高度和位置符合预期,但整体 K-mer 数量远超基因组大小的估算,且没有明显的杂合峰或重复峰。2)在比对文件中,这些 Reads 的起始和终止坐标完全重合。
假说 B:样本杂合性(Heterozygosity-induced Misalignment)
- 来源:样本基因组是杂合的,但参考基因组是纯合的。
- 逻辑:样本的两个等位基因 A1 和 A2 存在序列差异。如果参考基因组只包含 A1 的版本,A2 的 Reads 可能会被迫比对到 A1 的位置。
- 结果:在比对层面上表现为该位置的深度(Depth)异常或非特异性堆叠,被误认为是某种形式的冗余。
假说 C:参考基因组塌缩(Collapsed Repeats)
- 来源:样本基因组中有 10 个重复拷贝,但参考基因组由于组装问题只保留了 1 个。
- 逻辑:样本中来自 10 个物理位置的 Reads 都会比对到参考基因组的这 1 个点上。
- 结果:产生极高的“多对一”比对率。
另:PCR 偏好在 k-mer 分布中的具体体现及诊断证据
主峰的形态畸变:峰宽与过离散(Overdispersion)
在理想状态下,k-mer 频数应符合泊松分布,其方差等于均值(Var=μ)。
-
PCR 偏好的表现:由于不同区域的扩增效率不同,原本应该集中在深度 M 的 k-mer 被“打散”到了 0.5M到 1.5M 的区间内。
-
诊断证据:k-mer 主峰变得矮胖、宽阔,甚至失去明显的峰尖。在数学拟合中,这被称为过离散现象。
-
后果:这会导致 GenomeScope 等软件在计算时,残差(Residuals)变大,降低对杂合度和重复率估算的置信度。
GC 含量诱导的伪重复与伪缺失
PCR 酶对不同 GC 含量的序列具有化学选择性(通常偏向扩增 GC 含量中等的片段)。
-
假说 A(高 GC 偏好):若样本中某些区域 GC 含量适中,被过度扩增。
- k-mer 表现:这些区域的 k-mer 会向右移动,进入“重复序列”的高频区(2M,3M 层级)。这会产生伪重复(False Positives for Repeats)的假象。
-
假说 B(极端 GC 抑制):极高或极低 GC 区域扩增效率极低。
- k-mer 表现:这些区域的 k-mer 频数向左移动,掉入低频区(1−5×),甚至与测序错误峰重叠。这会导致基因组大小被低估。
K-mer 与 GC 含量的联合分布(GC-Content Plot)
要确证 k-mer 分布异常是否由 PCR 偏好引起,通常需要引入第二维证据:k-mer 频数 vs. GC 含量图。
-
诊断证据:
-
如果发现 k-mer 深度 随 GC 含量 呈现明显的抛物线关系(即 GC 在 45%−55% 时深度最高,两端骤降)。
-
结论:此时 k-mer 分布图中的“长尾”或“多峰”极有可能是 PCR 偏好造成的技术偏离,而非样本基因组真实的生物学重复。
-
综合判定方法:K-mer 与其他技术的结合
k-mer 分析虽然强大,但在处理空间结构和系统偏差时存在局限,需结合其他方法:
外部基准验证
-
C-value 查询:在实验前通过流式细胞术或数据库(如 Kew Garden, GenomeSize.com)获取预估基因组大小,用于校准 K-mer 计算结果。
-
$G = \frac{N_{total}}{M}$:利用 K-mer 总数除以主峰深度得到估算大小。若此值显著大于组装长度,提示存在大量重复序列塌缩。
污染与偏差检查
-
GC 分布图:检测是否存在 GC 偏好性。若 GC 含量极高,二代测序会产生大量碎片。
-
NT 库比对:通过随机抽取 Reads 进行 BLAST 比对,排除外源物种污染导致的高重复假象。
结构性验证
-
BAM 比对频率图:利用
Picard识别 PCR 冗余(技术重复),区分其与生物学重复序列。 -
BUSCO 评估:通过单拷贝基因的找回率判定基因组的完整性及是否存在多倍化情况。
常用软件工具
k-mer分析软件简介
k-mer分析分为k-mer频数统计和基因组特征评估两步。此外,Smudgeplot还可以用k-mer分析评估物种的倍性。
- jellyfish
- jellyfish可以实现第一步k-mer频数统计。
- 特点是使用Hash表存储数据,能多线程运行;速度快,内存消耗小。
- KMC
- KMC可以实现第一步k-mer频数统计。
- GenomeScope
- GenomeScope可以实现第二步,利用k-mer频数统计结果进行基因组特征评估。
- 1.0版本用于二倍体物种,2.0版本用于多倍体物种。
- KAT(The k-mer Analysis Toolkit)
- KAT(The k-mer Analysis Toolkit)可以实现k-mer频数统计和基因组特征评估两步。
- 包含多个工具来帮助用户通过使用k-mer对测序数据进行简单分析,如组装完整性、测序错误、是否有污染等。
- GCE
- GCE可以分别实现k-mer频数统计和基因组特征评估两步。
- KmerGenie
- KmerGenie可以同时实现k-mer频数统计和基因组特征评估两步。
- 最大优点在于可以实现在多个预设k-mer下的自动分析,除了进行常规的k-mer频数统计之外,还能够基于不同k-mer自动计算基因组大小,并为基因组组装评估一个最佳组装k-mer数值作为备选。
一些使用软件的经验总结
- 【推荐】用Smudgeplot评估物种倍性后,用组合jellyfish+GenomeScope1.0做二倍体物种的基因组调查,用组合KMC+GenomeScope2.0做多倍体物种的基因组调查。
- k-mer长度常用17/21。
- 软件KmerGenie,GCE和jellyfish获取的频数分布表,都可用于软件genomescope和GCE第二步骤的分析。
- 由于老版本的GCE第一步骤支持的最大k-mer频数为255,大于255的数据被合并(新版本GCE好像更新了,目前没有这个限制了);而jellyfish统计到10000行,预估结果会更为准确。
- GenomeScope对于高重复序列的基因组统计的基因组大小会偏小,建议max kmer coverage设置大一点,大于等于10000。
- 有些软件(比如GCE)有另一个参数需注意和设定,单倍体模式还是杂合模式,可以两种模式都分析,查看差别。
- 实践经验发现,k-mer值设置得越高,估计出来的基因组size会越大;
- 另外,在jellyfish里的jellyfish histo统计频数分布时,用参数-h 10000把统计上限调高,以及在GenomeScope阶段,Max kmer coverage设置的大一些(即统计进的kmer数量越多),估算出来的基因组大小也会略大一些。