本页面已设置访问密码,请输入密码查看全文:
摘要:核心种质(Core Set)是“从海量种质中挑出一个小而精、却能在遗传/表型空间上代表整体”的子集,本质是一个在给定成本约束下的多目标优化问题:在代表性(coverage)、内部多样性(diversity)以及业务结构约束(PC/MC 等)之间做权衡。本篇重点说明核心种质筛选的技术背景。
这个分析要解决什么问题
在实际项目里,我们经常会遇到这些场景:
- 种质库里有成百上千甚至上万份材料,但下游实验、深度测序或田间试验的预算只能覆盖几百份。
- 客户希望我们帮忙“先筛一批代表性强的材料”,后续这批材料会被频繁使用(功能研究、育种亲本、指纹图谱等)。
- 原始基因型数据质量参差不齐,如果不先梳理、压缩成一个结构清晰的小集合,后续分析很难稳定复用。
所以,核心种质筛选要解决的核心矛盾是:
- 样本太多:不可能对所有材料都做高成本、长周期的深度研究;
- 又不能随便删:乱删会丢掉关键变异、重要亚群或稀有等位基因,后面想补救就很难。
核心种质筛选做的事情,用一句不那么严谨的话说就是:
“在不把‘重要信息’丢掉的前提下,把样本数压到一个可操作的规模”。
 Source: https://www.blitzllama.com/blog/sampling-errors
Source: https://www.blitzllama.com/blog/sampling-errors
从数据视角看:核心种质筛选到底在干嘛
可以把这件事拆成三个直观的问题:
- 问题 1:数据长什么样?
- 通常我们会拿到 VCF/PLINK 或 SNP 矩阵,再配上样本的品种类型、地理来源、育种阶段、表型等信息。
- 第一步是做常规 QC:缺失率过滤、深度/HWE 检查、去掉明显异常样本,得到一个“稳定可用”的基因型矩阵。
- 问题 2:群体是什么结构?
- 用 PCA、亲缘关系矩阵、
ADMIXTURE等手段,看一下群体有没有明显的亚群、地理结构或批次效应。 - 这一步的结果,会直接影响后面“每个亚群要保留多少材料”这类决策。
- 用 PCA、亲缘关系矩阵、
- 问题 3:在这个基础上挑一批“既能代表整体,又不太冗余”的样本
- 直观理解:
- 不希望核心集中所有材料都挤在 PCA 图的某一个角落;
- 也不希望核心集内部高度重复(几乎是同一个基因型反复出现)。
- 为了达到这个效果,我们会用一些量化指标和专门的软件(如 Core Hunter 3)来帮忙做选择,而不是“肉眼挑样本”。
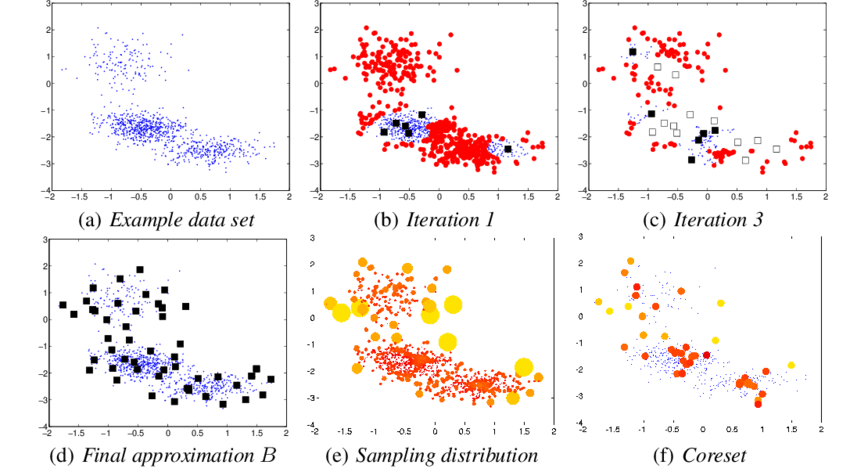 Source: Dan Feldman et al. 2011
Source: Dan Feldman et al. 2011
- 直观理解:
先把基因型矩阵整理干净 + 看清楚群体长相,再在这个空间里“画圈选代表”。
一些必须知道的技术概念(简化版)
- 种质资源(Germplasm Resources):携带可遗传信息、可用于研究与育种的生物材料集合。
- 核心种质(Core Set):从原始种质集合中抽取的一批“代表材料”,希望在更小规模下尽量保留总体的遗传/表型多样性。
- 代表性(Representativeness/Coverage):
- 核心集合对原始集合的覆盖程度;强调“原始集合里的每个个体/类型,都能被核心集合中的某个个体较好地代表”。
- 常见做法是看“全集中每个样本,到核心集中最近样本的距离”的平均值(如A-NE)。
- 距离越小,说明“全集里的样本,都能在核心集中找到一个挺像的邻居”,代表性就越好。
- 内部多样性(Internal Diversity):
- 核心集合内部个体彼此差异的程度。
- 希望核心集内部的样本彼此不要太相似,避免浪费配额。
- 实际上会用一些多样性指标(等位基因数目、稀有等位基因比例、多样性指数等)来衡量。
- 结构约束(让结果“可解释、可交付”):
- 很多项目会有类似“每个地理区域/品种类型都要有代表”的需求。
- 技术上可以通过设定“各亚群的大致占比”和“每个亚群至少要选几个”来实现(很多工具里用 PC/MC 这类术语表达)。
- DNA 指纹图谱(DNA Fingerprint Map):使用分子标记(常见为 SNP/SSR)为样本群体建立的“身份标识”,用于鉴定、保护与追溯。
选核心集时,不是只看一个指标,而是在“代表性、内部多样性和结构均衡”三件事之间做平衡。
一个典型的技术流程
- 整理输入数据
- 收集样本列表、元数据(地理来源、品种类型、育种阶段、表型等);
- 准备好对应的 VCF/PLINK/基因型矩阵和参考基因组信息。
- 做一轮必要的 QC 和结构分析
- 用
vcftools/PLINK过滤掉质量明显不合格的样本和位点; - 用 PCA/亲缘关系/
ADMIXTURE看清楚群体结构和亚群分布。
- 用
- 根据甲方要求确定“选谁”的基本原则
- 允许的核心集规模大致区间(比如先看 5%、10%、20%);
- 哪些亚群/地区/类型必须被覆盖,哪些可以适度压缩;
- 更看重“代表全集”还是“多样性最大化”,或者两者都要兼顾。
- 用专业工具跑出一个或一组候选核心集
- 常见工具包括
Core Hunter 3、GenoCore等; - 尝试不同规模、不同参数组合,看指标和结构表现的变化趋势。
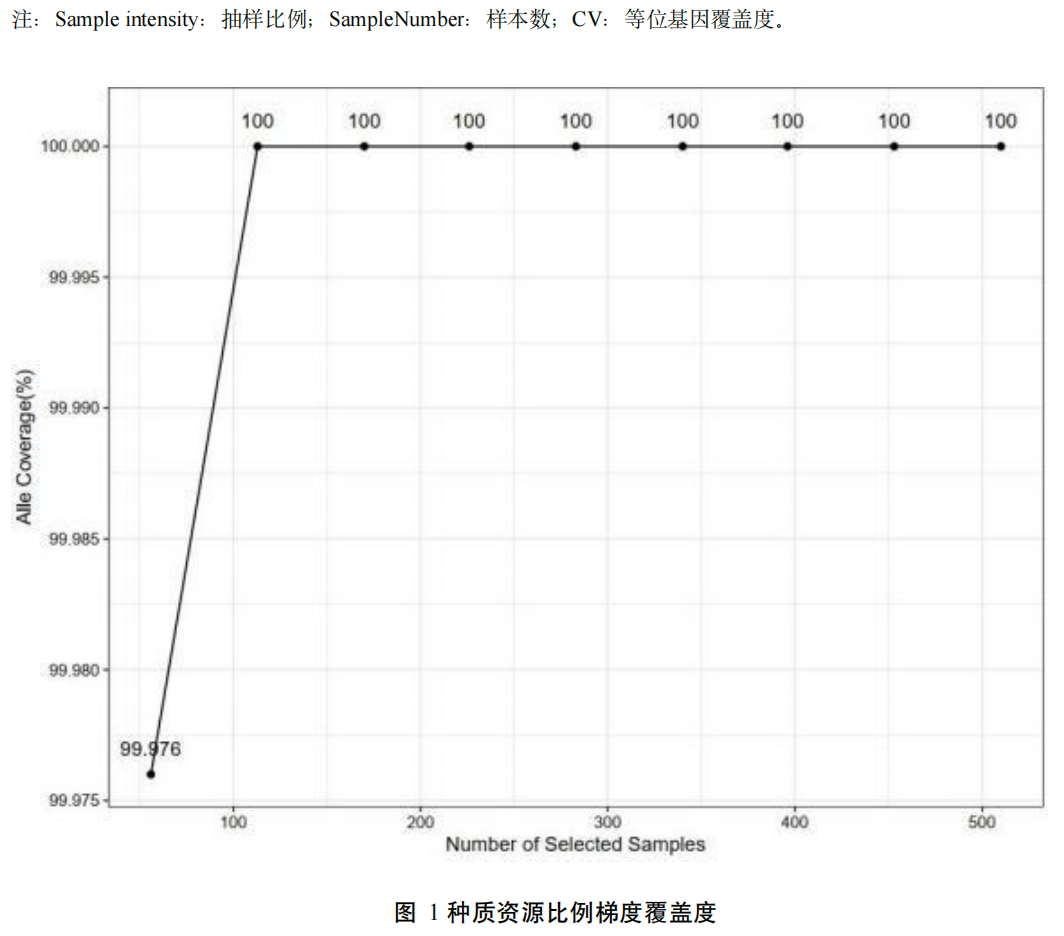
- 常见工具包括
- 对候选核心集做“体检”和可视化
- 对比核心集 vs 全集的多样性、代表性、结构覆盖情况;
- 用 PCA/树图/聚类图等方式,直观展示“有没有缩小样本但保留整体结构”。
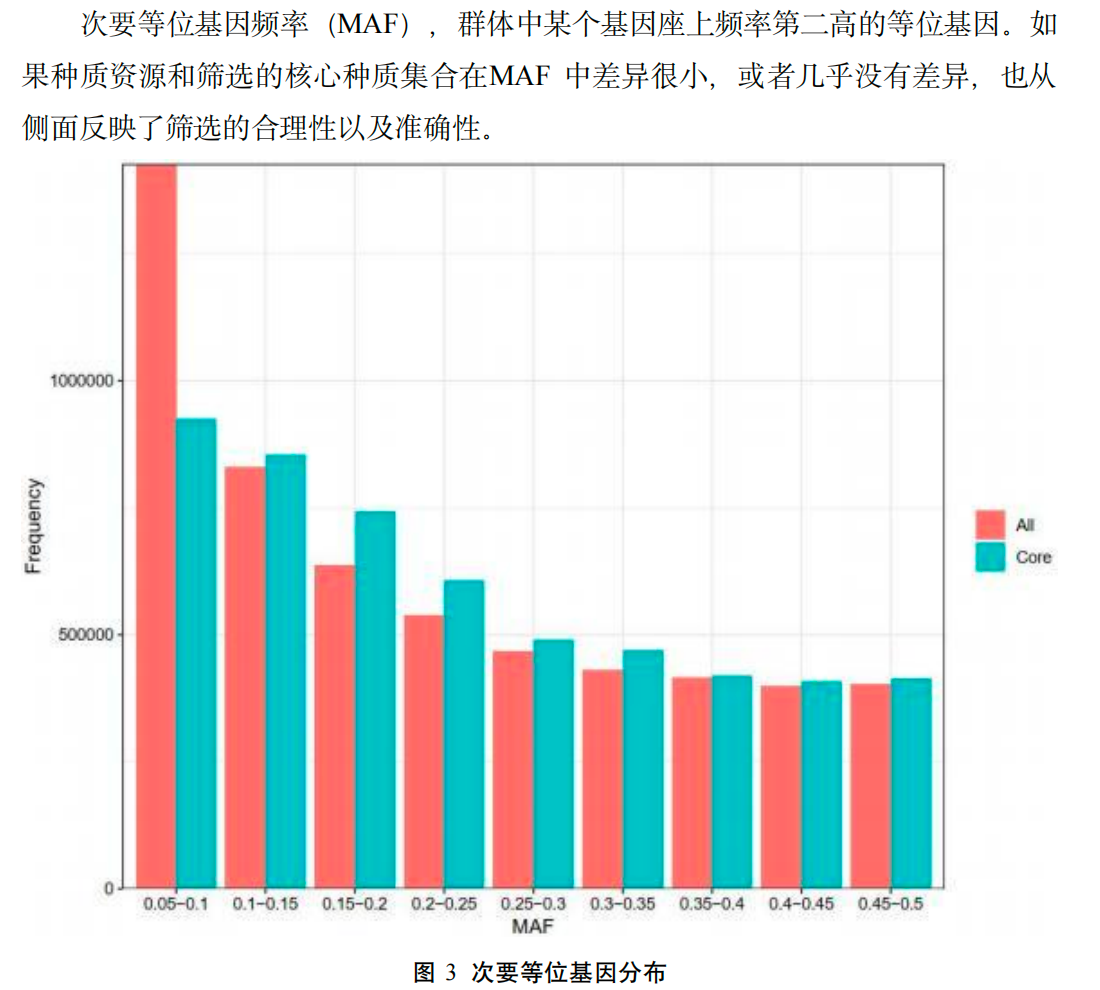
- 承接下游分析
- 固定住核心集名单、关键参数、工具版本和随机种子;
- 为后续 GWAS/QTL/GS、DNA 指纹图谱开发、资源库优化等分析打一个统一的数据基础。
以 DNA 指纹图谱 为例,可以粗略理解为:
- 从Coreset筛选出一小批质量高、分布均匀、多态性强、区分度高的代表性位点标记;
- 这些标记组合形成“指纹”,可以在同种内稳定地区分不同材料,且不受环境影响,适合用于品种鉴定、种子纯度检测、资源多样性研究和品种产权保护等场景。
核心种质筛选一般怎么被使用
在实际项目里,核心种质筛选很少是“做完就结束”的一次性分析,更常见的是作为一块可复用的基础组件,长期服务于不同的下游应用。大致可以分成四类典型用法:
- 面向科研与功能解析的“前置压缩”
- 先从大群体中筛出一批代表性材料,再在这批材料上做 GWAS/QTL/GS、转录组/代谢组、候选基因精细解析等。
- 目的:在可控样本规模内提升统计功效和发现效率,让后续复杂分析既“算得动”,又更容易解释与复现。
- 面向资源管理的“去冗余”
- 帮资源库管理方识别高度冗余的材料,以及“必须保留”的多样性代表。
- 核心集可以被视为当前阶段的“物种/群体基准面板”,用于指导入库、扩繁、更新和淘汰决策,降低长期维护成本。
- 面向品种鉴定与审定流程的“ID”
- 许多指纹图谱或 SNP/INDEL 位点面板项目,会优先在核心集中设计和评估标记,再将同一套位点扩展到更大规模的样本。
- 典型应用包括:品种真实身份鉴定、侵权与纠纷取证、种子/种苗质量追踪(二维码/条码化交付)等。
- 面向保护与长期监测的“核心监测群体”
- 对濒危物种、小群体或重要育种材料,核心集可以作为“重点跟踪的一批个体”,用于监测遗传多样性随时间的变化。
- 在保护遗传学/保护基因组学和泛基因组研究中,也常用核心集来覆盖主要结构单倍型/变异,为后续扩展测序和结构变异分析提供代表性样本框架。
友商产品情况
友商通常以“群体遗传分析报告 + 指纹图谱标准产品 + 位点开发/育种决策支持”的组合交付:
-
综合型服务商:强调从下机数据质控、变异检测、群体结构分析的一站式。 如美吉生物或诺禾致源将其作为群体遗传业务的其中一项服务。

-
垂直领域厂商:更强调把核心种质与功能基因组/育种决策(亲本选择、材料分层)绑定在一起。