这篇笔记整理 从零开始做一项GWAS 的通用流程,并把常见软件工具(示例:TASSEL、METAL等)与关键参数/注意点放在同一处,便于以后做实战时直接对照补全。
通用GWAS分析流程(从数据到信号)
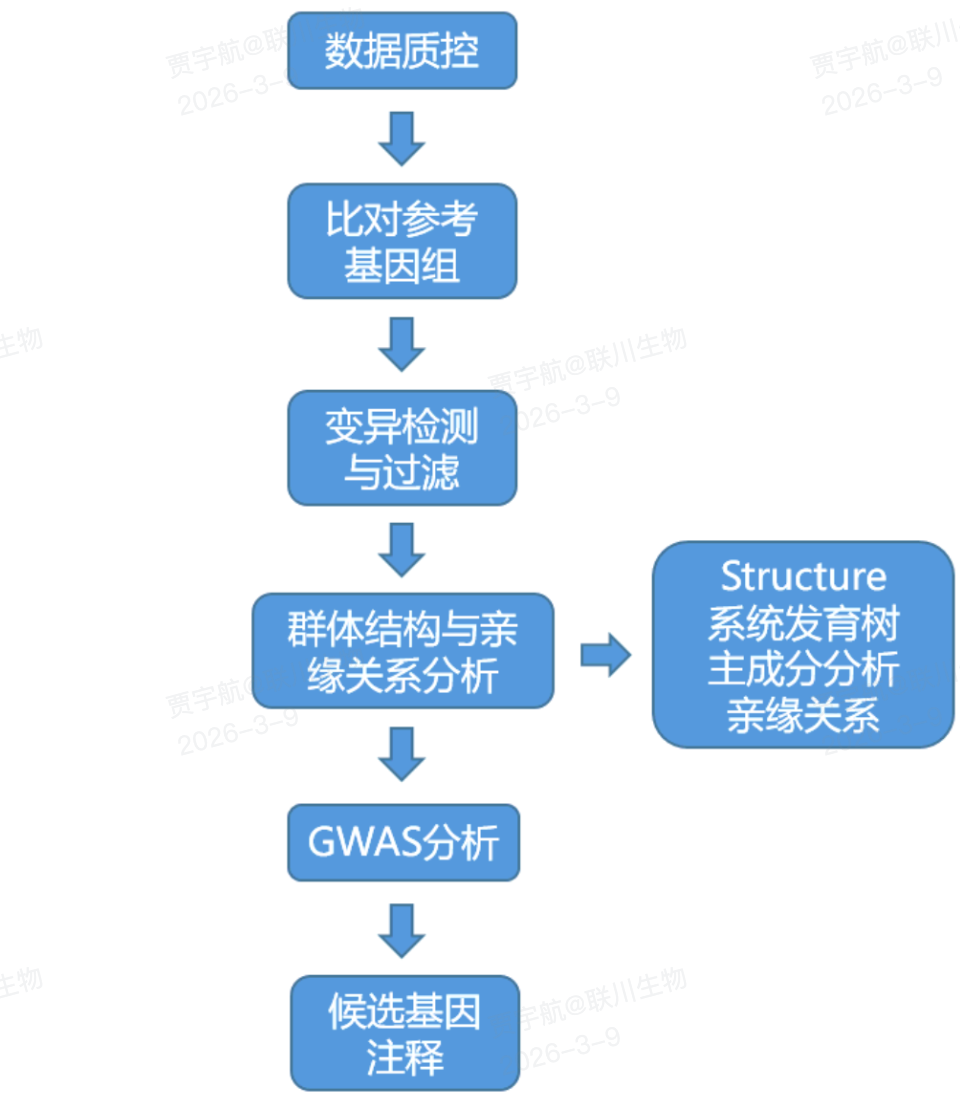
- 获取样本与表型信息(Phenotyping)
- 明确性状定义、测量方式、批次/环境因素
- 记录协变量:性别、年龄、批次、场地、测量人等(后续进入模型)
- 获取基因型数据(Genotyping)
- WGS / SNP芯片 / GBS / WES 等
- 变异与样本质量控制(QC)
- 位点层面:缺失率、MAF、HWE(视物种/群体而定)
- 样本层面:缺失率、性别核对、异常杂合度、重复/近亲
- 插补与过滤(可选但常见)
- 参考面板 + LD 进行基因型插补
- 插补后按 INFO/\(r^2\)/CR 等指标过滤低质量位点
- 群体结构/亲缘关系/LD分析
- PCA/祖先成分、亲缘关系矩阵(GRM/K矩阵)、LD衰减
- 目标:减少群体结构导致的假阳性
- 关联分析(Association testing)
- 线性模型/混合线性模型(定量性状)
- 二分类性状需用逻辑回归/广义线性混合模型等
- 结果质控与可视化
- QQ-plot(通胀/校正是否合理)
- Manhattan plot(定位信号峰)
- 信号整理与下游
- lead SNP、LD clumping、条件分析
- 候选基因与功能注释、精细定位、共定位/因果推断等
从零开始做GWAS(四个核心步骤)
- 样本收集与表型测定(Phenotyping):明确性状定义与协变量(性别、批次、环境等)
- 基因型测定(Genotyping):WGS/芯片等得到全基因组变异
- 统计学关联分析(Association testing):逐位点扫描,在线性/混合线性模型里检验 \(\beta\) 是否显著
- 确定GWAS显著信号:按阈值筛选 lead SNP/信号峰(并配合QQ/Manhattan检查结果合理性)
常见软件与它们在流程里的位置
- TASSEL
- 常用于植物/育种场景的GWAS工作流(可输出 Manhattan/QQ 等)
- 更适合作为“入门全流程”工具:从表型+基因型到关联与基础可视化
- METAL
- 用于 Meta-GWAS:把多个队列/群体的GWAS汇总统计进行合并
- 关键点:对齐效应等位基因、统一编码、处理反向链与等位基因频率
- SnpEff(下游)
- 用于变异功能注释:外显子/内含子/启动子/基因间区、同义/非同义等
关键参数/注意点清单(以后实战时补实参)
QC常见阈值(示例,需按数据/物种调整)
- 样本缺失率:如
--mind 0.05(PLINK语境) - 位点缺失率:如
--geno 0.05 - MAF:如
--maf 0.01或0.05 - HWE:按研究设计(自然群体/家系/强选择)选择是否启用与阈值
结构校正
- PCA协变量:常用前若干PC作为固定效应
- 亲缘关系矩阵:混合线性模型里作为随机效应,抑制家系结构假阳性
显著性阈值
- 常见全基因组阈值 \(5\times10^{-8}\)(人类)
- 其他物种/标记密度下常按有效检验数调整
“文献驱动”的流程例
GWAS数据处理与筛选流程(文字版)
- 收集与执行独立的GWAS:2,056个独立GWAS / 207个复杂经济性状
↓ - 使用 METAL 进行跨群体 Meta-GWAS:合并相同性状的独立GWAS结果
↓ - 生成 Meta-GWAS 汇总统计数据:共 268 个 Meta-GWAS 结果
↓ - 与 molQTL 变异位点重叠对齐:对齐到 3,087,268 个经过测试的高质量 SNPs
↓ - 显著性阈值筛选:保留 lead SNP 的 \(P < 1\times10^{-5}\) 位点
↓ - 得到显著 GWAS 位点:共 1,507 个,用于后续多组学整合分析
GWAS 与 molQTL 的整合解读(把“信号”连到“机制”)
这一类研究通常会在GWAS之后做“GWAS信号与分子QTL的重合/整合”,核心关注点可概括为:
- 富集(enrichment):GWAS信号是否在不同类型的molQTL中显著富集(如eQTL、sQTL等)
- 介导遗传率:复杂性状的遗传率有多少比例可被分子表型“介导/解释”(例如用MESC等方法)
- 四类互补整合策略(示例)
- 共定位(fastENLOC):判断分子表型与复杂性状是否共享潜在因果变异(如RCP阈值)
- 孟德尔随机化(SMR + HEIDI):推断分子表型到复杂性状的因果链条,并用HEIDI降低LD造成的假阳性
- 单组织TWAS(S-PrediXcan):评估某组织内预测表达与性状关联
- 多组织TWAS(S-MultiXcan):跨组织整合提高统计效力
- 组织相关性:用组织相关性得分等指标,定位与性状最相关的“核心组织”
- 互补性:不同类型molQTL对GWAS信号的解释往往互补;有些GWAS位点可能只能被某一类QTL(如sQTL、lncQTL)解释
整合分析框架(文字版)
- 数据输入
- GWAS:复杂性状GWAS汇总统计数据(207个性状 / 268个GWAS结果)
- molQTL:多组织molQTL图谱(34个组织的 eQTL / sQTL / eeQTL 等)
- 全局评估
- 位点重叠筛选(GWAS位点 ∩ molQTL位点)
- 富集分析:评估 GWAS 信号在不同 molQTL 中的富集
- 介导遗传率评估:例如 MESC
- 整合映射(互补策略)
- 共定位:fastENLOC(共享潜在因果变异)
- 孟德尔随机化:SMR + HEIDI(因果推断 + 降低LD假阳性)
- 单组织TWAS:S-PrediXcan
- 多组织TWAS:S-MultiXcan(整合提升效力)