这篇笔记整理了根据项目实战来学习variant calling(SNP和InDel)的流程。
总结
- 原始数据:FASTQ(PE150,20 个猪样本)
- 质量评估 & 接头去除(FastQC / fastp / Trimmomatic,关键指标:Q30、GC、dup rate)
- 比对到参考基因组(BWA-MEM / 其他,比对参数、参考基因组版本)
- 标记/去除 duplicates(Picard / samtools markdup,为什么要做、什么时候不能随便扔)
- 重校正(若有 BQSR:需要哪些 known sites)
- 单样本变异检测(GATK HaplotypeCaller 或其他 caller)
- 多样本联合分型(如 GVCF → GenotypeGVCFs)
- 变异过滤(hard filter / VQSR,常用阈值)
- 注释与下游分析(VEP / ANNOVAR / SnpEff 等)
开始之前的问题
- SNP calling的全流程,以及每一步的原理和可能的trouble是什么?
- SNP calling的操作过程中,都需要用到什么database,什么输入数据?
相关信息
参考资料:
- https://ming-lian.github.io/2019/02/08/call-snp/
目标
- 通过20个猪样本variant calling项目,理解该流程的结构、软件、参数和所有输出文件结构以及最终解读。通过实战来掌握其中的术语概念、原理、意义。达到能独立上手和讲解的水平。(践行费曼学习法)
- 该分析在最新的methods类文章都有哪些方面的讨论?
- 思考:如果面对的是大队列需求,怎么能缩短项目周期和节约资源?
基础概念
- Single Nucleotide Polymorphism (SNP):群体中特定位点上两种或多种不同的核苷酸(通常是转换Transition「同类碱基置换,如嘌呤之间AtoG或嘧啶之间CtoT」或颠换Transversion)存在,且在该群体中的等位基因频率通常高于 1%。该术语倾向于描述群体遗传学、物种进化或亲缘关系分析中的多态性,属于胚系变异。
- SNP calling:旨在通过高通量测序数据比对,识别个体基因组中与参考序列不同的单个碱基变异(SNP)。
- QTL:QTL的划定是一个基于统计学推断的过程,它没有全球统一的、绝对的物理范围或SNP数量标准,但有一套被广泛接受的科学方法和经验性阈值。详见[[02_QTL的判定]]
- expression quantitative trait loci (eQTLs):指基因组中能影响特定基因(mRNA或蛋白质)表达水平的遗传变异(通常为SNP)。它通过研究DNA变异与基因表达量的相关性,揭示遗传突变调控表达的机制,主要分为顺式(cis-eQTL,近距离,上下游1Mb)和反式(trans-eQTL,远距离不同染色体或相距>5Mb)两类。
- Genotype-Tissue Expression Project (GTEx数据库):是目前最全的人类组织特异性eQTL数据库。
- Genomics Reference Panel:一个包含了大量个体全基因组测序(WGS)数据的“高密度变异数据库”它记录了群体中极其丰富的自然遗传变异。
分析流程
质量控制和过滤
碱基质量评估方法
Q20:该碱基出错率0.01 Q30该碱基出错率0.001 Q=-10log(Pvalue)
详情见笔记[[03_双端测序接头识别与去除]]
SNP calling流程
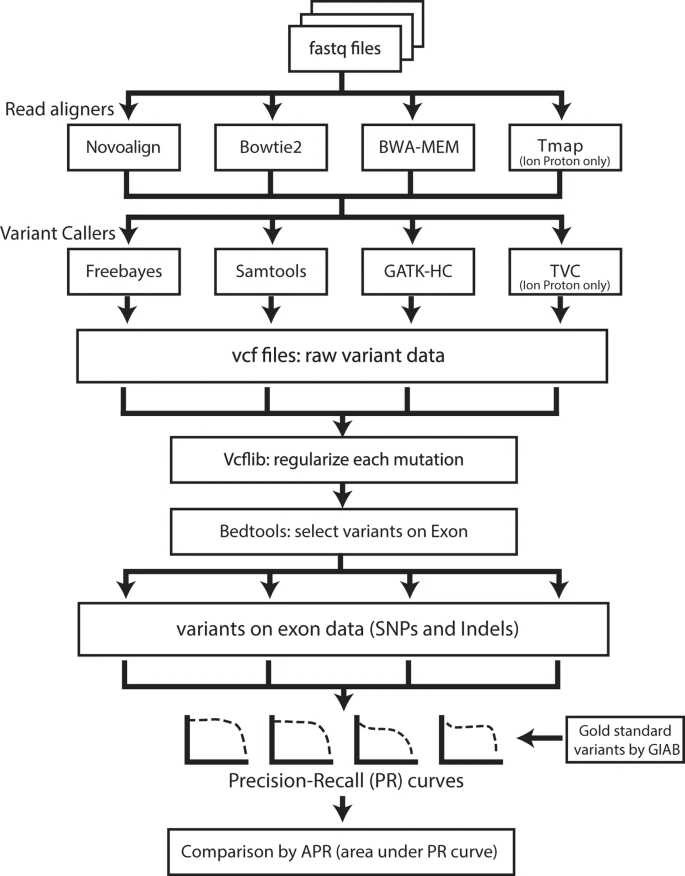
RNAseq based
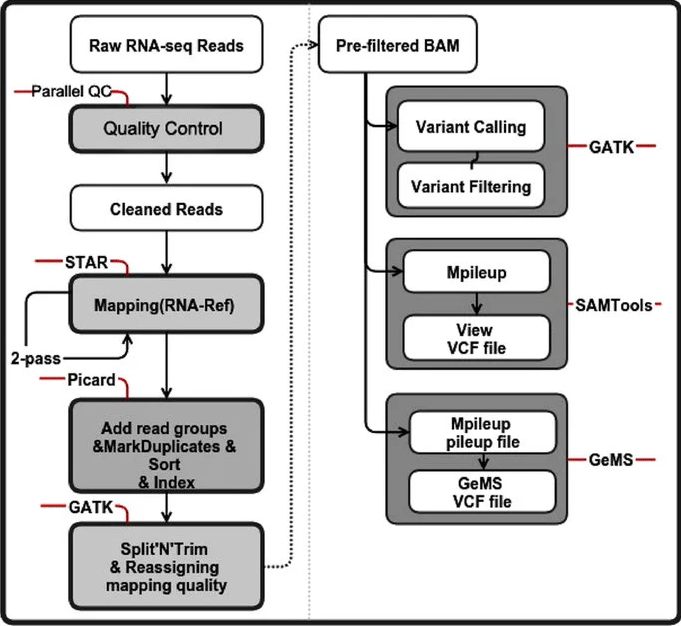
原始数据与项目背景
- 目的: 搞清楚样本设计与测序方案,为后续每一步设置合理阈值。
- 输入:
- 原始测序数据:
*.fastq.gz,通常为双端测序PE150。 - 元数据:每个样本的编号、批次、组织来源、表型信息等。
- 原始测序数据:
- 输出: 无新文件,主要是记录清楚样本信息表(建议放在
Summary/1_Data_Quality_Control/Data_Statistics.xlsx中)。 - 常见 trouble:
- 批次信息和文件命名不一致,后期很难追踪 → 一开始就固定命名规范。
- 样本/表型表缺字段,后面做 GWAS/pQTL 时需要返工补充。
原始读长质量控制(QC)与接头去除
- 目的: 去掉低质量碱基和接头污染,保证下游比对和变异检测可靠。
- 输入: 原始
FASTQ。 - 输出:
- QC 报告:FastQC / fastp 的
html报表,放在1_Data_Quality_Control/QC_Report。 - 过滤后的
clean FASTQ:命名如sample1.clean.R1.fastq.gz。
- QC 报告:FastQC / fastp 的
- 常用工具 & 关键参数示例:
- FastQC:整体质量评估。
- fastp(示意命令):
1 | |
- 关键指标:
- Q30 比例(>80% 一般可接受,>90% 较好)。
- GC 含量是否接近期望范围。
- duplicates 率是否异常偏高(结合下文
duplicates 的产生原因理解来源)。
- 常见 trouble & 对策:
- Q30 太低 → 检查测序批次质量,考虑更严格过滤或补测。
- 接头污染严重 → 调整接头识别参数或检查文库构建策略。
- duplicates 率偏高 → 可能是 PCR 轮次太多或上机文库浓度过高。
比对到参考基因组(Alignment)
- 目的: 将 clean reads 准确定位到参考基因组上,为后续变异检测准备
BAM。 - 输入: clean FASTQ;参考基因组
FASTA及其索引。 - 输出:
- 原始比对结果:
sample1.raw.bam(或中间sam)。 - 排序后的比对结果:
sample1.sorted.bam。
- 原始比对结果:
- 常用工具 & 命令示例(BWA-MEM):
1 | |
- 检查点:
- 比对率(overall alignment rate):WGS 通常应 >95%,太低要检查参考基因组是否匹配、是否有污染。
- 插入片段长度分布是否合理。
- 每条染色体/基因组整体深度分布是否符合预期(可视化输出到
2_Mapped_Statistics目录)。
- 常见 trouble & 对策:
- 比对率很低 → 可能是物种/版本错配、污染或建库失败。
- 某些染色体几乎无覆盖 → 检查参考基因组是否包含对应 contig / 染色体名是否一致。
标记/去除 duplicates
- 目的: 标记由 PCR/cluster/光学原因引起的重复 reads,避免在变异检测中放大证据。
- 输入:
sample.sorted.bam。 - 输出:
- 标记重复后的 BAM:
sample.markdup.bam。 - 对应索引:
sample.markdup.bam.bai。
- 标记重复后的 BAM:
- 常用工具 & 命令示例(samtools markdup 或 Picard):
1 | |
- 概念关联: 这里用到的 duplicates 类型详见文末
## duplicates的产生原因。 - 检查点:
- duplicates 率:视建库和物种而定,一般 5–20% 较常见;若 >30–40% 需要结合文库策略解释。
- 常见 trouble:
- 全部去除 duplicates 会降低有效深度,尤其是低起始量或高扩增文库 → 某些分析只标记不删除。
基因组重校正(BQSR,可选)
- 目的: 利用已知变异位点校正系统性测序偏差,提高变异检测准确性。
- 输入:
sample.markdup.bam;已知高质量变异集(known sites)。 - 输出: BQSR 后的 BAM:
sample.bqsr.bam。 - 适用性:
- 人类/模式生物有成熟 known sites(如 dbSNP、1000G)时强烈推荐。
- 某些家畜物种(如猪)可根据是否有高质量面板酌情选择;若没有可靠 known sites,可考虑跳过 BQSR 并在笔记中说明理由。
单样本变异检测(Single-sample Calling)
- 目的: 在每个样本上识别潜在 SNP/InDel。
- 输入: 对每个样本的最终 BAM(标记 duplicates,视情况 BQSR)。
- 输出:
- 若走 GVCF 流程:每个样本一个
sample.g.vcf.gz。 - 或直接得到
sample.vcf.gz(不推荐用于大队列联合分析)。
- 若走 GVCF 流程:每个样本一个
- 工具示例(GATK HaplotypeCaller):
1 | |
- 检查点:
- 每样本 SNP 数量是否在合理范围。
- 深度分布、GQ 分布是否正常。
多样本联合分型(Joint Genotyping)
- 目的: 在所有样本的 GVCF 上进行联合建模,得到统一的 cohort VCF。
- 输入: 所有样本的
*.g.vcf.gz。 - 输出:
- 联合分型后的
cohort.raw.vcf.gz(包含所有样本的基因型)。
- 联合分型后的
- 工具示例:
1 | |
- 检查点:
- 变异位点总数与预期是否一致。
- 每个位点的缺失率、等位基因数。
变异过滤与质控(Filtering)
- 目的: 去除低可信度位点,保证后续关联分析的统计可靠性。
- 输入:
cohort.raw.vcf.gz。 - 输出:
- 过滤后的高质量变异:
cohort.filtered.vcf.gz。 - 可选:额外导出
plink格式或矩阵用于下游分析。
- 过滤后的高质量变异:
- 过滤方式:
- Hard filter: 手动设置阈值(常用于样本数不极大的项目)。
- VQSR: 需要训练集和资源较多。
(详细的字段与阈值见下文“VCF 关键字段与过滤规则”。)
变异注释与结果整理
- 目的: 将纯“坐标层面”的变异转化为“生物学信息”:哪些基因、哪些功能后果。
- 输入:
cohort.filtered.vcf.gz;注释数据库(Ensembl VEP cache / ANNOVAR DB / SnpEff DB 等)。 - 输出:
- 注释过的 VCF 或表格文件,例如
cohort.filtered.ann.vcf.gz或.txt/.xlsx。 - 统计汇总表:功能类别、转录本影响、等位基因频率分布等。
- 注释过的 VCF 或表格文件,例如
- 用途:
- 后续的 GWAS/eQTL/pQTL 关联分析。
- 与已知功能变异库、ClinVar 或物种特异数据库比对。
交付物
1 | |
VCF 关键字段与过滤规则
- 常看字段(INFO / FORMAT):
DP:测序深度(depth)。过低的位点(如 DP < 5–8)可靠性差。QD:Variant Confidence / Depth。过低(如 QD < 2.0)常被认为是假阳性。FS:Fisher Strand,衡量正负链偏倚。偏倚严重(如 FS > 60)可疑。MQ:比对质量均值(mapping quality)。极低 MQ 表明比对不可靠。MQRankSum:比对质量在 REF/ALT 之间是否有显著差异。ReadPosRankSum:REF/ALT 在 read 上的位置分布是否有显著差异。GQ:每个样本的基因型置信度。
- 示例 Hard Filter 规则(需根据项目微调):
1 | |
- 复习时 checklist:
- 看总体 SNP/InDel 数量是否在合理区间。
- 抽查几个位点在 IGV 中表现是否与 VCF 记录一致。
- 记录本项目最终实际采用的阈值,并在 methods 中保持一致。
后续关联分析 (Association)
-
基因型 (Genotype): 通过 SNP Calling 获得每个样本在特定位点的等位基因状态(如 0, 1, 2,代表突变个数)。
-
表型 (Phenotype): 这里的性状是分子的“量”,例如蛋白质丰度。
统计模型会对全基因组的 SNP 逐一进行检验。
-
原假设 (H0): 该 SNP 与蛋白质丰度无关(相关系数 β=0)。
-
备择假说 (H1): 该 SNP 的基因型剂量变化会导致蛋白质丰度发生显著变化。
详情见[[02_GWAS关联分析]]
后续功能分类与证据评估
-
Cis-pQTL: 显著 SNP 物理位置靠近编码基因。这提供了较强的先验概率,暗示其可能直接影响转录或翻译。
-
Trans-pQTL: 显著 SNP 远离编码基因。
-
诊断性评估: 此时需要评估是否存在连锁不平衡 (LD)。即:该 SNP 是真正的功能位点,还是仅仅与功能位点距离太近而被“带”出来的信号?
duplicates的产生原因
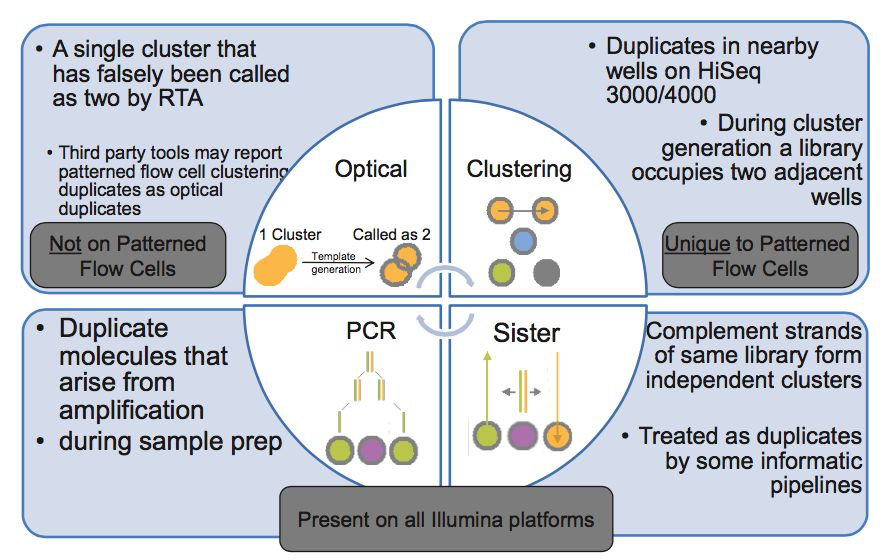
PCR duplicates(PCR重复)
PCR扩增时,同一个DNA片段会产生多个相同的拷贝,第4步测序的时候,这些来源于同一个拷贝的DNA片段会结合到Fellowcell的不同位置上,生成完全相同的测序cluster,然后被测序出来,这些相同的序列就是duplicate
Cluster duplicates
生成测序cluster的时候,某一个cluster中的DNA序列可能搭到旁边的另一个cluster的生成位点上,又再重新长成一个相同的cluster,这也是序列duplicate的另一个来源,这个现象在Illumina HiSeq4000之后的Flowcell中会有这类Cluster duplicates
Optical duplicates(光学重复)
某些cluster在测序的时候,捕获的荧光亮点由于光波的衍射,导致形状出现重影(如同近视散光一样),导致它可能会被当成两个荧光点来处理。这也会被读出为两条完全相同的reads
Sister duplicates
它是文库分子的两条互补链同时都与Flowcell上的引物结合分别形成了各自的cluster被测序,最后产生的这对reads是完全反向互补的。比对到参考基因组时,也分别在正负链的相同位置上,在有些分析中也会被认为是一种duplicates。