这篇笔记整理了在做基因组注释与变异检测时,底层文件格式的含义。
主要覆盖参考基因组构建(BWA 索引、功能注释)、Fastq 原始测序数据、以及 VCF 变异结果文件中的关键字段。
参考基因组
1 | |
01_Build_Genome_DB.sh: 核心构建脚本。包含从原始数据下载、过滤、索引构建到功能提取的全流程命令。nohup.out: 记录脚本运行过程中的标准输出与错误日志,用于追溯构建过程中的异常。02_raw: 存储从公共数据库(如 Ensembl)下载的原始未处理文件。.gff3: 原始基因组注释。.dna_sm.toplevel.fa: 软掩盖(Soft-masked)的基因组序列,重复序列被标记为小写字母。
03_genome:存储处理后的序列及各类访问索引。*.fa.gz(cds, gene, mrna, pep, promoter): 提取出的各类功能元件序列包。genome.fa/genome.gff: 经过标准化处理(如重命名染色体)的主参考文件。- BWA 索引 (.amb, .ann, .bwt, .pac, .sa): 用于二代测序数据比对。
- 访问索引 (.fai, .dict): 用于随机定位与校验。
Ref/:ref.genome.*: 该子目录下文件通常为 downstream 流程(如 GATK)专用。id_table.txt: 关键对照表,记录原始染色体 ID 到标准化 ID 的映射。
stat/:gene@mrna//ncrna_gene@lnc_rna/等: 按“父特征@子特征”命名的分类统计。name/: 包含 ID 与基因/转录本名称的映射(name_gene.txt等)。description/: 存储基因的功能描述信息,用于注释补全。table_per_feature_type.txt: 该类特征的数量、平均长度等统计报表。
stat_features.txt: 整个基因组所有特征类型的总览统计。
04_diamond:使用 DIAMOND 工具将提取的 CDS/mRNA 序列与蛋白库进行比对。-
*.go.txt/*.kegg.txt/*.nr.txt: 初始比对结果,记录了查询序列与数据库序列的相似性得分(E-value, Identity)。
mRNA2gene.txt: 关系映射表,用于将比对到的转录本功能归类到所属基因上。
-
05_annotation:将比对结果经过滤、去重、权重筛选后生成的最终注释表。GO.annotation.txt: 基因与 Gene Ontology 词条的对应关系。KEGG.annotation.txt: 基因与代谢通路(Pathways)的对应关系。NR.annotation.txt: 基因与已知蛋白功能的详细文本描述对照表。
测序原始文件
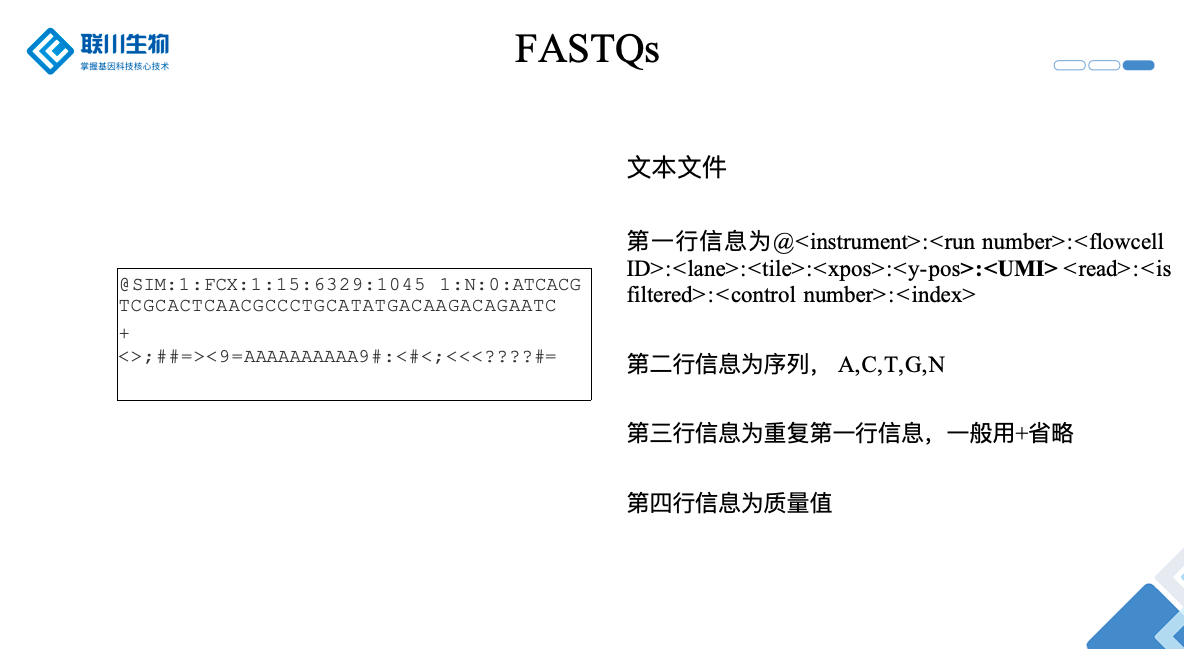
原始数据质控
fastp质控输出
*_CleanData_R1.fastq.gz & *_CleanData_R2.fastq.gz
fastp统计 json/html
整合所有样本的报告 data_stat.txt
fastQC质控报告
对 CleanData 进行,输出zip/html报告。
序列访问索引
这类索引用于实现对大型 FASTA 文件的随机访问(Random Access),使软件能够快速定位并提取特定染色体或位置的碱基。
samtools索引
samtools faidx ref.genome.fa
该指令会生成genome.fa.fai。
 注:某些性染色体(如 Y 染色体)或部分高度重复区域的组装结果可能比某些长 Scaffold 还要短。仅凭长度排序
注:某些性染色体(如 Y 染色体)或部分高度重复区域的组装结果可能比某些长 Scaffold 还要短。仅凭长度排序sort -k2nr是一种启发式搜索,不能作为最终证据。建议结合第一列的 NAME 命名规范进行二次判定。
基于BWA的比对索引
bwa index -a bwtsw ref.genome.fa
通常不需要关注这些文件结构。
| 扩展名 | 格式/内容解释 |
|---|---|
| .bwt | Burrows-Wheeler Transform:核心索引,将序列转换为数学表示。 |
| .ann | Annotation:记录染色体 ID、长度及在 .pac 中的连接顺序。 |
| .amb | Ambiguous:记录非 ATGC 碱基(如 N)的位置。 |
| .pac | Packed:二进制。将原始碱基以 2-bit 形式压缩存储。 |
| .sa | Suffix Array:后缀数组采样点,用于坐标还原。 |
.amb:
# 头部行(第一行):参考基因组的总长度,参考序列的数量(染色体或 Contig 的总数),内部校验值或特定格式标记
# N区域起始位置,N区域长度,N区域所在的序列名或索引(通常与.ann文件对应)
2501912388 613 544
90927422 100 n
91945429 100 n
92881119 100 n
214753360 100 n
......
.ann:
# 头部行(第一行):参考基因组的总长度,参考序列的数量(染色体或 Contig 的总数),内部校验值或特定格式标记
# 序列注释块两行为一组:第一行分别为,该序列在整个连接参考序列中的起始偏移量(Offset),序列名称,该序列的描述信息。
# 第二行分别为, 内部标记(通常标明该序列在链接过程中的某些状态),该序列的精确长度,该序列包含的非变体或特定字符(如N)的数量类别标识。
2501912388 613 11
0 1 (null)
0 274330532 4
0 2 (null)
274330532 151935994 6
0 3 (null)
426266526 132848913 10
0 4 (null)
559115439 130910915 5
0 5 (null)
......
picard索引
$java -Xmx$picard_mem -jar $picard CreateSequenceDictionary R=$ref_db O=$dict && touch $DONE/picard_index.done
生成ref.genome.dict:

.dict 文件由两部分组成:
@HD(Header Line):定义文件版本和排序方式。@SQ(Sequence Dictionary Line):定义参考基因组中每一条染色体或 Contig 的具体信息。
每列意义:
| 标签 (Tag) | 名称 | 意义 | 示例/备注 |
|---|---|---|---|
@HD |
Header | 文件的头部起始标志 | 表明这是一个头部行 |
VN |
Version | 格式版本号 | 1.5 |
@SQ |
Sequence | 序列记录起始标志 | 每一行代表一条染色体或 Contig |
SN |
Sequence Name | 序列名称 | 如 1, 2, X 或 Contig ID FPKY02000001.1 |
LN |
Length | 序列长度 | 单位为碱基对 (bp)。例如染色体 1 长度为 274330532 |
M5 |
MD5 Checksum | 序列内容的 MD5 校验码 | 用于唯一标识序列内容,防止因文件名相同但内容不同导致的错误 |
UR |
URI | 参考基因组源文件的路径 | 指向原始 .fa 文件的位置,通常为绝对或相对路径 |
比对过程文件
*.bam
sam文件的压缩二进制表示:
 Header 头部信息:以
Header 头部信息:以 SAM 文本格式存储的元数据,包含版本、序列字典(@SQ)、读段分组(@RG)和比对工具(@PG)。在 BAM 中,这部分被转换为二进制存储。
Alignments比对部分:
| 列号 | 字段名称 | 含义说明 | 示例/取值范围 |
|---|---|---|---|
| 1 | QNAME | 序列 ID (Query Name) | 来自测序仪的原始 ID |
| 2 | FLAG | 数字标识 (Bitwise Flag) | 代表比对状态的位运算总和 |
| 3 | RNAME | 参考序列名称 (Reference Name) | 染色体编号,若未比对上则为 * |
| 4 | POS | 比对起始位置 (Position) | 基于 1 的最左端位置 |
| 5 | MAPQ | 比对质量值 (Mapping Quality) | 比对结果错误的概率(−10log10P) |
| 6 | CIGAR | 简并比对表示法 | 描述 Match、Mismatch 及 Indel |
| 7 | RNEXT | 配对端比对到的染色体 | = 表示与本 Read 相同,* 表示无配对 |
| 8 | PNEXT | 配对端比对到的位置 | 配对 Read 的起始位置 |
| 9 | TLEN | 观察到的片段长度 (Template Length) | 插入片段大小,通常左端为正,右端为负 |
| 10 | SEQ | Read 的碱基序列 | A, T, C, G, N |
| 11 | QUAL | 碱基质量得分 (Phred Quality) | ASCII 编码的碱基准确性信息 |
FLAG
FLAG 列通过一个十进制数字存储了多个布尔状态(True/False)。它是通过将以下二进制位累加得到的。
常用标志位(部分):
1 (0x1): 该 Read 是双端测序(Paired-end)。2 (0x2): 该 Read 每一端都正确比对上(Properly aligned)。4 (0x4): 该 Read 未比对上(Unmapped)。16 (0x10): 该 Read 比对到了负链(Reverse strand)。256 (0x100): 二次比对(Secondary alignment)。1024 (0x400): 重复记录(PCR or Optical Duplicate)。
例:FLAG 为 99,其二进制分解为 64+32+2+1:
- 1: 双端测序
- 2: 正确匹配
- 32: 配对端在负链
- 64: 这是双端中的第一条 Read (R1)
- (蕴含信息:本条 Read 在正链)
MAPQ:比对质量
MAPQ 是评估比对结果“唯一性”的关键指标,计算公式为:
MAPQ=$−10log_{10}P(mapping\ is\ wrong)$
- 40 - 60: 高置信度,Read 极大概率只属于当前位置。
- 0: 极低置信度。通常表示 Read 序列过于简单(如全 A)或在基因组中存在多个几乎相同的拷贝,软件无法确定其真实位置。
CIGAR:比对状态描述
CIGAR 字符串告诉我们 Read 是如何“贴”在参考序列上的:
- M: Alignment Match (可以是 Mismatch,仅表示比对位置对齐)。
- I: Insertion (相对于参考基因组,Read 多出了碱基)。
- D: Deletion (相对于参考基因组,Read 缺失了碱基)。
- S: Soft Clipping (末端未比对上的部分,但在记录中保留了碱基序列)。
- H: Hard Clipping (末端未比对上的部分,且在记录中删除了该段序列)。
可选标签 (Optional Tags)
在 11 列之后,通常会有形如 NM:i:1 或 MD:Z:100 的标记。
1 | |
异常检测建议
观察到某行的数据不符合上述逻辑,可以采用以下排查思路:
- 若 FLAG 包含 4(未比对):则 POS 必须为 0,CIGAR 必须为
*。若不符,说明文件在生成过程中可能存在逻辑错误或软件 Bug。 - 若 TLEN 远大于文库长度:可能存在染色体结构变异(SV),或者该 Read 的两端被错误地比对到了相隔极远的位置。
- 线性对齐:一条read比对到参考序列上,可以存在插入(insert)、缺失(delete)、跳跃(skip)、剪切(clip),但是方向不变(不能是一部分和正链匹配,另一部分又和负链匹配),sam文件中只占用一行记录。
- 嵌合比对:由于一条测序read比对到基因组上时分别比对到两个不同的区域,而这两个区域基本没有接触和重叠。因此它在sam文件中需要占用多行记录显示。只有第一个记录称作”representative”,其他的都是”supplementary”。RNA-seq中的chimeric read或许可以说明有融合基因存在,但在基因组中一般作为结构变异的证据。
*.sort.bam
比对并按坐标排序后的二进制文件。
Picard MarkDuplicates
*.rmdup.bam
经过 Picard (CPU) 或 Parabricks (GPU) 处理,标记了 PCR/Optical 重复后的文件。
FLAG 标签
普通 BAM 文件中的 Read 通常没有关于重复的信息。而在 rmdup.bam 中,被识别为重复的 Read,其 FLAG 列数值会增加 1024 (0x400)。
-
普通比对 Read (R1, 正向比对): FLAG = 99
-
标记为重复后的相同 Read: FLAG = 1123 (99+1024)
“标记”与“删除”的策略差异
根据运行参数 REMOVE_DUPLICATES 的不同,结果有两种表现:
-
默认 (False):保留所有 Read,仅通过 FLAG 位进行标记。下游工具(如 GATK
HaplotypeCaller)会识别此 FLAG 并主动忽略这些数据。 -
删除 (True):物理删除被识别为重复的 Read。此时文件体积会显著减小,但该操作不可逆,通常不建议在临床或高标准研究中使用。
坐标排序要求
MarkDuplicates 运行前要求 BAM 文件必须按坐标(Coordinate)或查询名(Queryname)排序,因此 rmdup.bam通常继承了严格的排序属性。
*.metrics
.metrics 文件是一个以制表符分隔(Tab-delimited)的文本文件,记录了文库的复杂度和重复率统计信息。它通常分为三个主要部分:
命令行信息 (Header)
包含运行 Picard 的版本、具体参数设置以及处理时间。
核心指标表 (METRICS CLASS)
这是分析的核心,定义了文库的质量特征。
| 字段名称 | 含义 |
|---|---|
| LIBRARY | 文库名称(来源于 BAM 头的 @RG 标签)。 |
| UNPAIRED_READS_EXAMINED | 检查的单端 Read 总数。 |
| READ_PAIRS_EXAMINED | 检查的双端 Read 对总数。 |
| SECONDARY_OR_SUPPLEMENTARY_RDS | 次要或补充比对的 Read 数量。 |
| UNMAPPED_READS | 未比对上的 Read 数量。 |
| UNPAIRED_READ_DUPLICATES | 标记为重复的单端 Read 数量。 |
| READ_PAIR_DUPLICATES | 标记为重复的双端 Read 对数量。 |
| PERCENT_DUPLICATION | 重复率。计算公式为:Duplicates/Examined Reads。 |
| ESTIMATED_LIBRARY_SIZE | 预估文库规模。基于现有数据推测出的文库中唯一分子的近似总数。 |
直方图数据
如果设置了相关参数,文件末尾会包含一组数据,用于描述随着测序深度的增加,发现新(唯一)分子的效率。常用于判断继续增加测序量是否还有意义。
如何解读高重复率?
当 .metrics 显示 PERCENT_DUPLICATION 过高(例如 > 20%)时,应考虑以下替代假说并寻找诊断性证据:
假说 A:测序量过度(Over-sequencing)
解释:文库本身的分子种类有限,但测序深度过高,导致同一个分子被反复测序。
诊断性证据:ESTIMATED_LIBRARY_SIZE 远小于总测序量。增加测序量后,重复率线性上升。
假说 B:起始 DNA 量不足或文库复杂度低
解释:实验起始的 DNA 模板太少,PCR 扩增次数过多以补偿浓度。
诊断性证据:即使总 Read 数不多,重复率依然很高;且 ESTIMATED_LIBRARY_SIZE 的绝对数值较低。
假说 C:光学重复(Optical Duplicates)
解释:测序仪拍照或信号聚类过程中的技术偏差。
诊断性证据:查看 READ_PAIR_OPTICAL_DUPLICATES 字段。若该值比例异常高,通常与测序芯片(Flowcell)载量或聚类算法有关。
比对后统计文件
Picard CollectInsertSizeMetrics
*.InsertSizeMetrics.txt
记录插入片段均值与标准差。
## htsjdk.samtools.metrics.StringHeader
# CollectInsertSizeMetrics HISTOGRAM_FILE=../Output/InsertSize_result/Sample_1/Sample_1.InsertSizePlot.pdf INPUT=../Output/Sample_1/Sample_1.rmdup.bam OUTPUT=../Output/InsertSize_result/Sample_1/Sample_1.InsertSizeMetrics.txt VALIDATION_STRINGENCY=LENIENT DEVIATIONS=10.0 MINIMUM_PCT=0.05 METRIC_ACCUMULATION_LEVEL=[ALL_READS] INCLUDE_DUPLICATES=false ASSUME_SORTED=true STOP_AFTER=0 VERBOSITY=INFO QUIET=false COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json USE_JDK_DEFLATER=false USE_JDK_INFLATER=false
## htsjdk.samtools.metrics.StringHeader
# Started on: Thu Jan 22 12:37:09 UTC 2026
## METRICS CLASS picard.analysis.InsertSizeMetrics
MEDIAN_INSERT_SIZE MODE_INSERT_SIZE MEDIAN_ABSOLUTE_DEVIATION MIN_INSERT_SIZE MAX_INSERT_SIZE MEAN_INSERT_SIZE STANDARD_DEVIATION R
EAD_PAIRS PAIR_ORIENTATION WIDTH_OF_10_PERCENT WIDTH_OF_20_PERCENT WIDTH_OF_30_PERCENT WIDTH_OF_40_PERCENT WIDTH_OF_50_PERCENT WI
DTH_OF_60_PERCENT WIDTH_OF_70_PERCENT WIDTH_OF_80_PERCENT WIDTH_OF_90_PERCENT WIDTH_OF_95_PERCENT WIDTH_OF_99_PERCENT SAMPLE LIBRARY REA
D_GROUP
293 289 42 2 267253735 296.417202 73.607692 79130765 FR 17 33 49 67 85 109 135 1
73 235 303 583
## HISTOGRAM java.lang.Integer
insert_size All_Reads.fr_count
2 226
3 224
4 287
5 268
......
*.InsertSizePlot.pdf
- 诊断假说: 若分布非单峰正态,可能是 DNA 片段化不均或磁珠筛选失效。

samtools flagstat输出每个样本的比对统计。
*_aln_stat.txt186234779 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 secondary 1688865 + 0 supplementary 21718577 + 0 duplicates 185728377 + 0 mapped (99.73% : N/A) 184545914 + 0 paired in sequencing 92272957 + 0 read1 92272957 + 0 read2 177551800 + 0 properly paired (96.21% : N/A) 183900400 + 0 with itself and mate mapped 139112 + 0 singletons (0.08% : N/A) 3380244 + 0 with mate mapped to a different chr 2409003 + 0 with mate mapped to a different chr (mapQ>=5) -
*.flagstat: 比对率统计(Mapped, Properly paired, Singleton)。 *.depth: 碱基水平的深度文件。由samtools depth生成,是depth_stat.pl计算覆盖度的基础。
BAM 索引与整体对齐统计汇总
*.bai
BAM 索引文件,允许 IGV 等软件快速查看特定位点。
变异检测过程文件
*.realign.bam
UnifiedGenotyper 模式下经过 InDel 局部重比对,消除 InDel 末端比对错位造成伪 SNP 的干扰。
Variant calling结果文件
VCF:

| 字段名 | 格式/内容解释 |
|---|---|
| CHROM | 变异所在的染色体 ID(如“1”、“chrX”) |
| POS | 变异在染色体上的物理位置(从 1 开始,单位为 bp) |
| ID | 变异的唯一标识符(如 dbSNP 编号“rsID”;若无则为“.”) |
| REF | 参考基因组的碱基(或序列),即未发生变异时的等位基因 |
| ALT | 变异等位基因(Alternative),与 REF 不同的碱基(或序列);多等位用逗号分隔 |
| QUAL | 变异位点的质量分数(Phred Scale),表示对该变异存在的置信度,值越高越可靠 |
| FILTER | 过滤结果,“PASS” 表示通过所有标准,其他为未通过的过滤标签或原因 |
| INFO | 逗号分隔的附加注释信息,如变异类型(SNP/INDEL)、功能注释、等位基因频率、深度等参数;具体内容取决于分析流程 |
| FORMAT | 后续样本信息格式。描述每个样本的字段顺序与类型(如 GT:DP 表示基型和测序深度) |
| SAMPLE1 | 按 FORMAT 顺序展示该样本的各项指标(如“0/1:26”,分别表示基因型0/1和深度26) |
| SAMPLE2 | 同上,代表下一个样本的数据 |
INFO列详细解读
从“变异检测(Calling)”到“质量过滤(Filtering)”再到“功能注释(Annotation)”,每个阶段产生的 VCF,其 INFO 列的侧重点存在显著的诊断性差异。
原始检测阶段 (Raw Variant Calling)
目标:评估变异信号是否真实存在,排查测序噪声(Artifacts)。
产生的 VCF:单样本 VCF 或 gVCF(Genomic VCF)。
INFO 核心关注点:测序深度分布、碱基质量偏差、链偏好性。
联合分型与群体评估阶段 (Joint Calling & Population Stats)
目标:在群体层面评估等位基因频率,通过群体特征修正个体位点的置信度。
产生的 VCF:多样本(Multisample)VCF。
INFO 核心关注点:等位基因计数(AC)、频率(AF)、群体层面的质量指标。
功能过滤与临床/育种注释阶段 (Filtering & Annotation)
目标:判定变异的生物学效应及其与表型的关联。
产生的 VCF:过滤后的 VCF、注释后的 VCF(Annotated VCF)。
INFO 核心关注点:基因组位置(内含子/外显子)、氨基酸改变、有害性预测分值。
-
INFO 字段分类解析
-
测序与比对质量指标(用于 Hard Filtering)
这些指标是区分真实变异与测序/比对误差的关键诊断依据。
| 字段 | 全称 | 诊断意义(贝叶斯视角下的排他性证据) |
|---|---|---|
| QD | QualByDepth | 变异置信度与深度的比值。若 QD 过低,说明高质量碱基支撑不足,可能是由重复序列区域的比对错误引起的。 |
| FS | FisherStrand | Fisher 链偏好性评分。若值过高(>60),说明变异仅出现在正向链或反向链中,高度疑似测序偏好造成的假阳性。 |
| SOR | StrandOddsRatio | 另一种衡量链偏好性的指标,尤其在深度较高时比 FS 更稳健。 |
| MQ | RMSMappingQuality | 比对质量均方根。若 MQ < 40,说明支持该变异的 Read 大多来自比对不明确的区域(如转座子、重复序列)。 |
| MQRankSum | MQ Rank Sum | 比较参考基因组 Read 与变异 Read 的比对质量差异。若差异显著,说明变异 Read 可能源自错配。 |
- 群体遗传学指标(用于群体研究/GWAS)
当样本量增加时,这些指标可作为评估位点多态性的证据。
| 字段 | 全称 | 说明 |
| AC | Allele Count | 该位点 ALT(变异)等位基因在当前群体中出现的总次数。 |
| AN | Allele Number | 该位点所有有效样本(非缺失)中等位基因的总数(二倍体通常为 2× 样本数)。 |
| AF | Allele Frequency | 等位基因频率 (AF=AC/AN)。在罕见疾病分析中,AF > 1% 通常被视为中性变异的排除证据。 |
- 功能注释指标(由 SnpEff/VEP 等工具添加)
这是将技术数据转化为业务结论的关键字段。
| 字段 | 说明 | 示例内容 |
|---|---|---|
| ANN / CSQ | 功能预测 (Consequence) | 记录变异对蛋白质的影响:missense_variant (错义), synonymous_variant (同义), stop_gained (提前终止)。 |
| LOF | 功能缺失 (Loss of Function) | 标识变异是否导致基因功能丧失。在育种中,这是筛选关键控制基因的核心证据。 |
| NMD | 无义介导降解 | 预测转录本是否会因为提前终止密码子而被细胞机制降解。 |
- 特殊格式:gVCF 里的 INFO 差异
如果运行的是 HaplotypeCaller 的 -ERC GVCF 模式,
-
END 字段:由于 gVCF 会记录“非变异区域(Non-variant blocks)”,END 表示该块状区域(Block)的结束位置。
-
逻辑变化:在 gVCF 阶段,INFO 并不提供复杂的变异指标,而是记录该区域作为“纯合参考”的置信度。这是为了在后续多样本 GenotypeGVCFs 阶段提供先验概率。
其它字段的理解
-
QUAL 为
.是什么意思? -
QUAL是位点层面的质量值(Phred),但在不少场景会缺失并写成.,常见原因包括: -
该 VCF 来自联合分型/二次处理流程,作者选择不输出或无法定义统一的 site-level QUAL;
-
该记录不是标准“变异位点”的评分(例如某些 gVCF 的非变异记录/中间格式转换)。
-
实战中更常用的是
FILTER、INFO(如QD/FS/MQ)和样本级FORMAT(如GQ/DP/AD/PL)综合判断。 -
FORMAT 常见会有哪些字段?怎么和每个 SAMPLE 对上?
-
FORMAT用冒号把“样本列里每一段的含义”写清楚,样本列按同样的冒号顺序给出值。 -
常见
FORMAT子字段(不同 caller/流程会略有差异): -
GT:基因型(genotype),最关键。 -
DP:该样本在该位点的测序深度(depth)。 -
AD:allelic depth,常见为REF,ALT1,ALT2...的计数,例如AD=10,3表示 REF 支持 10、ALT1 支持 3。 -
GQ:genotype quality(Phred),表示该样本基因型判定的置信度。 -
PL:phred-scaled likelihoods,基因型似然(通常是一串逗号分隔的整数,用于下游再推断)。 -
PS:phase set(相位集合 ID),配合GT中的|使用(不是所有 VCF 都有)。 -
例子:若
FORMAT=GT:AD:DP:GQ,则样本列形如0/1:10,3:13:45,逐段对应GT=0/1、AD=10,3、DP=13、GQ=45。 -
GT 里的
1/2、0|1、1|0、1|1怎么读? -
GT的数字是对REF/ALT的索引编号: -
0表示REF -
1表示ALT列里第 1 个等位基因 -
2表示ALT列里第 2 个等位基因(如果ALT是多等位,比如ALT=AT,CT) -
` / ` vs ` `: -
0/1:未定相(unphased)的杂合,表示两个染色体拷贝分别为 REF 和 ALT1,但不知道哪条单倍型上是哪一个。 -
0|1:已定相(phased),表示这两份等位基因的相位已知(哪条单倍型是 0,哪条是 1)。 -
常见组合的直觉:
-
0/0:纯合参考(无变异)。 -
0/1:杂合(一个 REF、一个 ALT1)。 -
1/1:纯合 ALT1(两个拷贝都是第 1 个 ALT)。 -
1/2:多等位杂合(一个拷贝是 ALT1、另一个拷贝是 ALT2;要求ALT至少有两个备选等位基因)。 0|1vs1|0:两者都表示杂合且已定相,但哪条单倍型携带 ALT 不同;如果下游做连锁/单倍型分析,这个方向信息会影响结果。