这篇笔记整理了在做基因组注释与变异检测时,常见目录结构与底层文件格式的含义。
主要覆盖参考基因组构建(BWA 索引、功能注释)、Fastq 原始测序数据、以及 VCF 变异结果文件中的关键字段。
参考基因组
1 | |
01_Build_Genome_DB.sh: 核心构建脚本。包含从原始数据下载、过滤、索引构建到功能提取的全流程命令。nohup.out: 记录脚本运行过程中的标准输出与错误日志,用于追溯构建过程中的异常。02_raw: 存储从公共数据库(如 Ensembl)下载的原始未处理文件。.gff3: 原始基因组注释。.dna_sm.toplevel.fa: 软掩盖(Soft-masked)的基因组序列,重复序列被标记为小写字母。
03_genome:存储处理后的序列及各类访问索引。*.fa.gz(cds, gene, mrna, pep, promoter): 提取出的各类功能元件序列包。genome.fa/genome.gff: 经过标准化处理(如重命名染色体)的主参考文件。- BWA 索引 (.amb, .ann, .bwt, .pac, .sa): 用于二代测序数据比对。
- 访问索引 (.fai, .dict): 用于随机定位与校验。
Ref/:ref.genome.*: 该子目录下文件通常为 downstream 流程(如 GATK)专用。id_table.txt: 关键对照表,记录原始染色体 ID 到标准化 ID 的映射。
stat/:gene@mrna//ncrna_gene@lnc_rna/等: 按“父特征@子特征”命名的分类统计。name/: 包含 ID 与基因/转录本名称的映射(name_gene.txt等)。description/: 存储基因的功能描述信息,用于注释补全。table_per_feature_type.txt: 该类特征的数量、平均长度等统计报表。
stat_features.txt: 整个基因组所有特征类型的总览统计。
04_diamond:使用 DIAMOND 工具将提取的 CDS/mRNA 序列与蛋白库进行比对。-
*.go.txt/*.kegg.txt/*.nr.txt: 初始比对结果,记录了查询序列与数据库序列的相似性得分(E-value, Identity)。
mRNA2gene.txt: 关系映射表,用于将比对到的转录本功能归类到所属基因上。
-
05_annotation:将比对结果经过滤、去重、权重筛选后生成的最终注释表。GO.annotation.txt: 基因与 Gene Ontology 词条的对应关系。KEGG.annotation.txt: 基因与代谢通路(Pathways)的对应关系。NR.annotation.txt: 基因与已知蛋白功能的详细文本描述对照表。
序列访问索引
这类索引用于实现对大型 FASTA 文件的随机访问(Random Access),使软件能够快速定位并提取特定染色体或位置的碱基。
genome.fa.fai
 注:某些性染色体(如 Y 染色体)或部分高度重复区域的组装结果可能比某些长 Scaffold 还要短。仅凭长度排序是一种启发式搜索,不能作为最终证据。建议结合第一列的 NAME 命名规范进行二次判定。
注:某些性染色体(如 Y 染色体)或部分高度重复区域的组装结果可能比某些长 Scaffold 还要短。仅凭长度排序是一种启发式搜索,不能作为最终证据。建议结合第一列的 NAME 命名规范进行二次判定。
基于BWA的比对索引
| 扩展名 | 格式/内容解释 |
| — | — |
| .bwt | Burrows-Wheeler Transform:核心索引,将序列转换为数学表示。 |
| .ann | Annotation:记录染色体 ID、长度及在 .pac 中的连接顺序。 |
| .amb | Ambiguous:记录非 ATGC 碱基(如 N)的位置。 |
| .pac | Packed:二进制。将原始碱基以 2-bit 形式压缩存储。 |
| .sa | Suffix Array:后缀数组采样点,用于坐标还原。 |
测序原始文件
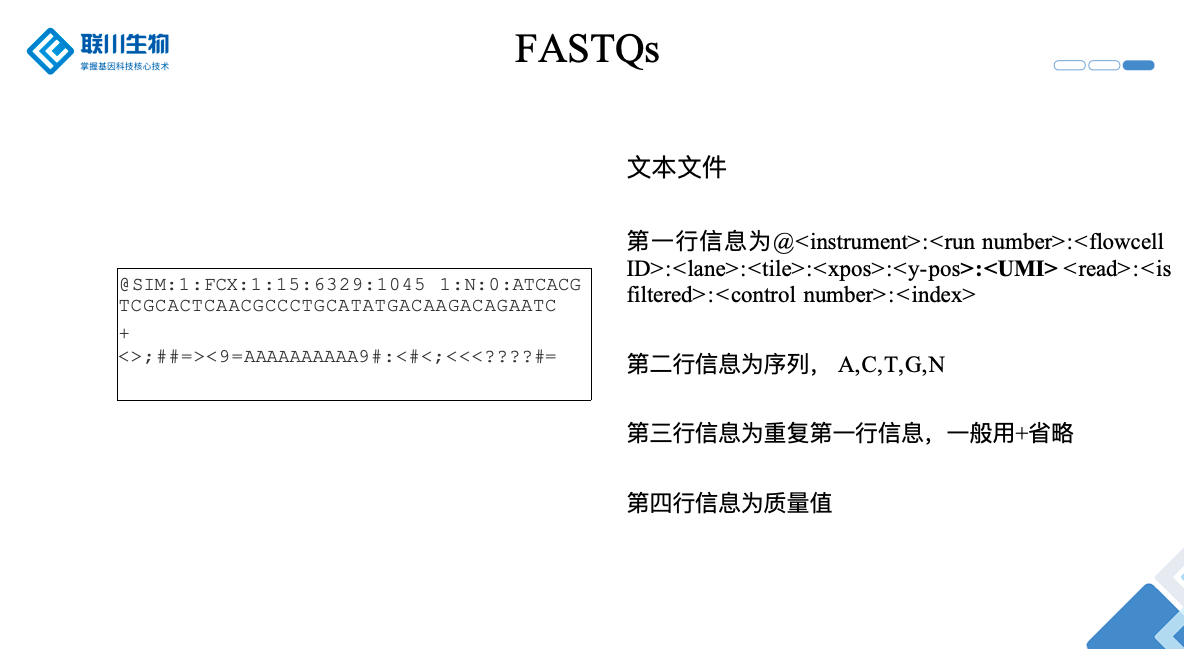
Variant calling结果文件
VCF

| 字段名 | 格式/内容解释 |
|---|---|
| CHROM | 变异所在的染色体 ID(如“1”、“chrX”) |
| POS | 变异在染色体上的物理位置(从 1 开始,单位为 bp) |
| ID | 变异的唯一标识符(如 dbSNP 编号“rsID”;若无则为“.”) |
| REF | 参考基因组的碱基(或序列),即未发生变异时的等位基因 |
| ALT | 变异等位基因(Alternative),与 REF 不同的碱基(或序列);多等位用逗号分隔 |
| QUAL | 变异位点的质量分数(Phred Scale),表示对该变异存在的置信度,值越高越可靠 |
| FILTER | 过滤结果,“PASS” 表示通过所有标准,其他为未通过的过滤标签或原因 |
| INFO | 逗号分隔的附加注释信息,如变异类型(SNP/INDEL)、功能注释、等位基因频率、深度等参数;具体内容取决于分析流程 |
| FORMAT | 后续样本信息格式。描述每个样本的字段顺序与类型(如 GT:DP 表示基型和测序深度) |
| SAMPLE1 | 按 FORMAT 顺序展示该样本的各项指标(如“0/1:26”,分别表示基因型0/1和深度26) |
| SAMPLE2 | 同上,代表下一个样本的数据 |
从“变异检测(Calling)”到“质量过滤(Filtering)”再到“功能注释(Annotation)”,每个阶段产生的 VCF,其 INFO 列的侧重点存在显著的诊断性差异。
原始检测阶段 (Raw Variant Calling)
目标:评估变异信号是否真实存在,排查测序噪声(Artifacts)。
产生的 VCF:单样本 VCF 或 gVCF(Genomic VCF)。
INFO 核心关注点:测序深度分布、碱基质量偏差、链偏好性。
联合分型与群体评估阶段 (Joint Calling & Population Stats)
目标:在群体层面评估等位基因频率,通过群体特征修正个体位点的置信度。
产生的 VCF:多样本(Multisample)VCF。
INFO 核心关注点:等位基因计数(AC)、频率(AF)、群体层面的质量指标。
功能过滤与临床/育种注释阶段 (Filtering & Annotation)
目标:判定变异的生物学效应及其与表型的关联。
产生的 VCF:过滤后的 VCF、注释后的 VCF(Annotated VCF)。
INFO 核心关注点:基因组位置(内含子/外显子)、氨基酸改变、有害性预测分值。
具体字段的理解
- QUAL 为
.是什么意思?QUAL是位点层面的质量值(Phred),但在不少场景会缺失并写成.,常见原因包括:- 该 VCF 来自联合分型/二次处理流程,作者选择不输出或无法定义统一的 site-level QUAL;
- 该记录不是标准“变异位点”的评分(例如某些 gVCF 的非变异记录/中间格式转换)。
- 实战中更常用的是
FILTER、INFO(如QD/FS/MQ)和样本级FORMAT(如GQ/DP/AD/PL)综合判断。
-
INFO 字段分类解析
- 测序与比对质量指标(用于 Hard Filtering) 这些指标是区分真实变异与测序/比对误差的关键诊断依据。
| 字段 | 全称 | 诊断意义(贝叶斯视角下的排他性证据) |
|---|---|---|
| QD | QualByDepth | 变异置信度与深度的比值。若 QD 过低,说明高质量碱基支撑不足,可能是由重复序列区域的比对错误引起的。 |
| FS | FisherStrand | Fisher 链偏好性评分。若值过高(>60),说明变异仅出现在正向链或反向链中,高度疑似测序偏好造成的假阳性。 |
| SOR | StrandOddsRatio | 另一种衡量链偏好性的指标,尤其在深度较高时比 FS 更稳健。 |
| MQ | RMSMappingQuality | 比对质量均方根。若 MQ < 40,说明支持该变异的 Read 大多来自比对不明确的区域(如转座子、重复序列)。 |
| MQRankSum | MQ Rank Sum | 比较参考基因组 Read 与变异 Read 的比对质量差异。若差异显著,说明变异 Read 可能源自错配。 |
1 | |
| 字段 | 全称 | 说明 |
|---|---|---|
| AC | Allele Count | 该位点 ALT(变异)等位基因在当前群体中出现的总次数。 |
| AN | Allele Number | 该位点所有有效样本(非缺失)中等位基因的总数(二倍体通常为 2× 样本数)。 |
| AF | Allele Frequency | 等位基因频率 (AF=AC/AN)。在罕见疾病分析中,AF > 1% 通常被视为中性变异的排除证据。 |
1 | |
- FORMAT 常见会有哪些字段?怎么和每个 SAMPLE 对上?
FORMAT用冒号把“样本列里每一段的含义”写清楚,样本列按同样的冒号顺序给出值。- 常见
FORMAT子字段(不同 caller/流程会略有差异):GT:基因型(genotype),最关键。DP:该样本在该位点的测序深度(depth)。AD:allelic depth,常见为REF,ALT1,ALT2...的计数,例如AD=10,3表示 REF 支持 10、ALT1 支持 3。GQ:genotype quality(Phred),表示该样本基因型判定的置信度。PL:phred-scaled likelihoods,基因型似然(通常是一串逗号分隔的整数,用于下游再推断)。PS:phase set(相位集合 ID),配合GT中的|使用(不是所有 VCF 都有)。
- 例子:若
FORMAT=GT:AD:DP:GQ,则样本列形如0/1:10,3:13:45,逐段对应GT=0/1、AD=10,3、DP=13、GQ=45。
- GT 里的
1/2、0|1、1|0、1|1怎么读?GT的数字是对REF/ALT的索引编号:0表示REF1表示ALT列里第 1 个等位基因2表示ALT列里第 2 个等位基因(如果ALT是多等位,比如ALT=AT,CT)
-
` / ` vs ` `: 0/1:未定相(unphased)的杂合,表示两个染色体拷贝分别为 REF 和 ALT1,但不知道哪条单倍型上是哪一个。0|1:已定相(phased),表示这两份等位基因的相位已知(哪条单倍型是 0,哪条是 1)。
- 常见组合的直觉:
0/0:纯合参考(无变异)。0/1:杂合(一个 REF、一个 ALT1)。1/1:纯合 ALT1(两个拷贝都是第 1 个 ALT)。1/2:多等位杂合(一个拷贝是 ALT1、另一个拷贝是 ALT2;要求ALT至少有两个备选等位基因)。0|1vs1|0:两者都表示杂合且已定相,但哪条单倍型携带 ALT 不同;如果下游做连锁/单倍型分析,这个方向信息会影响结果。