这篇笔记整理了QTL的鉴定原理。
[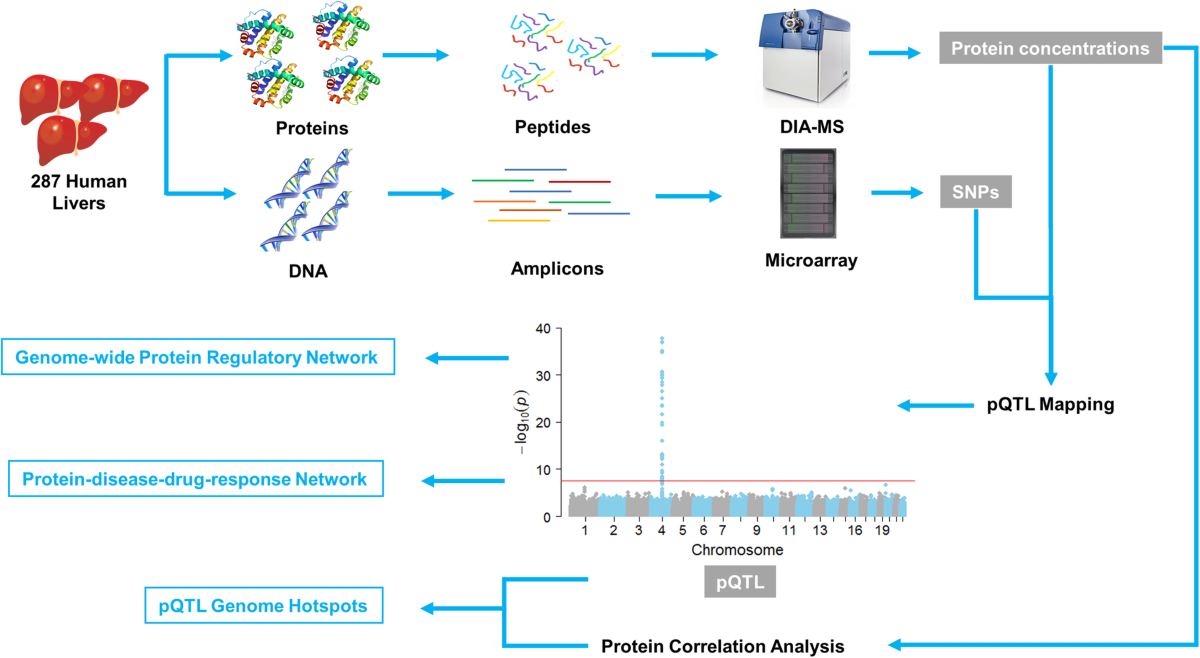 Bing He et. al. 2020
Bing He et. al. 2020
流程
- 全基因组关联扫描
- 对每个SNP标记(或每个基因组位置)进行统计检验,计算其与目标性状的关联强度。最常用的指标是 LOD值(对数优势比)。
- LOD值 Logarithm off the odds score:衡量该位点存在QTL的可能性比不存在QTL的可能性大多少倍的对数值。LOD值越高,关联越显著。
- 确定显著性阈值
- 这是第一个关键标准。 需要设定一个LOD阈值,超过该阈值的峰值区域才被认为是显著的QTL。
- 如何设定? 通常通过 “置换检验” 来确定。即:随机打乱性状数据与基因型数据的对应关系,重复扫描成千上万次,得到一个在零假设(无QTL)下的LOD值分布,然后取该分布的上分位数(如95%或99%)作为经验性显著性阈值。这能有效控制假阳性。
- 划定QTL区间(支持区间)
- 对于一个超过显著性阈值的LOD峰,其下的基因组区域就是一个候选QTL。但我们需要划定它的边界。
- 最常用方法:2-LOD支持区间。 从LOD峰值点向两侧下降,直到LOD值下降2个单位(即强度降为1/100)时,所对应的基因组位置就是该QTL的置信区间。这个区间有大约95%的概率包含真正的QTL。
- 另一个方法:贝叶斯可信区间。 用统计模型直接计算QTL位置的后验概率分布,然后取一定概率(如95%)的区间。
- 其他辅助判断标准
- 连续标记的支持: 一个可靠的QTL通常由多个相邻的、关联显著的SNP所支持,形成一个“峰”,而不是一个孤立的点。
- 表型变异解释率: 每个QTL能解释多大比例的表型变异(PVE)。研究中常将PVE > 10%的QTL称为“主效QTL”,这有助于聚焦最重要的遗传区域。
菊花研究为例进行说明:
High-density genetic map construction and identification of loci controlling flower-type traits in Chrysanthemum 在Song(2020)的文章中,作者很可能这样操作:
- 他们使用6452个SNP标记,对CTMD和RNRF性状进行了全基因组QTL扫描。
- 通过置换检验(或其他方法)确定了 LOD显著性阈值(例如,可能是3.0或更高)。只有LOD峰超过此阈值的区域才被报告为QTL。
- 对于每个显著的LOD峰,他们使用 2-LOD支持区间法 划定该QTL的物理范围(例如,位于LG1染色体上从20.5 cM到25.8 cM的区间)。
- 他们列出了每个QTL的 PVE值,并特别指出了PVE > 10%的“主效QTL”(如控制CTMD的3个主效QTL和控制RNRF的4个主效QTL)。
总结
- 没有固定的物理范围或SNP数量标准。 一个QTL的区间可能小到几kb(靠近单个基因),也可能大到几Mb(包含多个基因),这取决于重组事件、标记密度和QTL本身的效应大小。
- 核心标准是统计学的: 显著性阈值(LOD) 决定它是否存在,支持区间(如2-LOD drop) 决定它大概在哪里。
- 最终划定是一个结合统计结果和生物学知识的判断过程。 研究者会综合LOD峰形、支持区间、PVE以及区间内候选基因的功能等信息,来最终界定和报告QTL。
因此,QTL划定是遗传图谱分析中一项严谨的统计推断工作,其标准是灵活但具有明确统计学意义的