记录了建库以及上机前的原理。
测序样品的准备
DNA抽提检测
- 磁珠法(自动化程度高)、柱式法(快速纯度高)或传统的苯酚/氯仿法(适合超大片段提取)。
- 样本要求: 血液、唾液、组织、细胞等需无污染,并确保DNA的完整性(特别是大分子量DNA对长读长测序至关重要)。
- 质量控制(QC): 使用Qubit测量浓度,Agarose Gel Electrophoresis或TapeStation/Bioanalyzer分析完整性(RIN值)。
文库构建
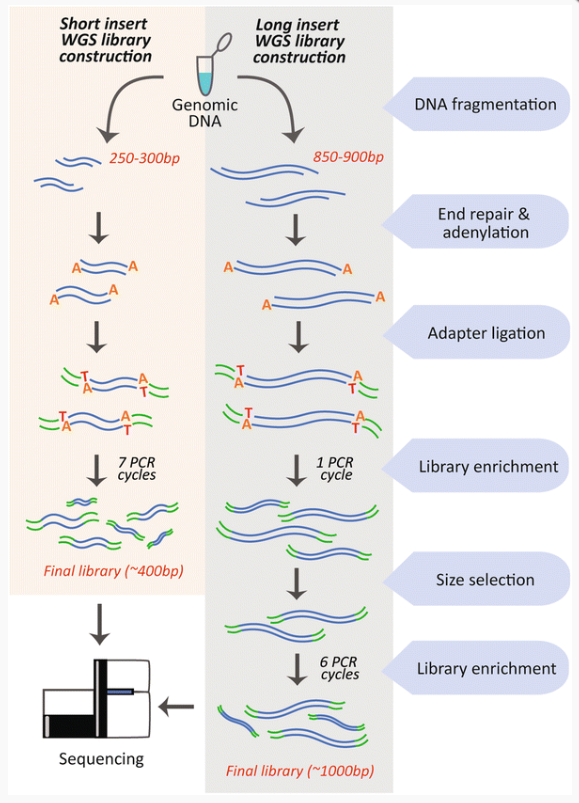 Jonathan J. Keats et. al. 2018.
Jonathan J. Keats et. al. 2018.
构建结果,每条read的状态如下:
P5−[i5 Index]−Read 1 SP−(Insert)−Read 2 SP−[i7 Index]−P7
- P5/P7 (Adapter):负责结合 Flow Cell 芯片表面。
- i5/i7 (Index/Barcode):位于接头与引物结合位点之间,负责标记样本。
- Read 1/2 SP (Sequencing Primer binding site):测序引物结合位点。

注:
- 酶切根据GC含量和预期片段密度选择限制性内切酶;酶切后需要产生特定的overhangs。通过凝胶电泳可判断DNA是否变为弥散的Smear条带图谱。
- GC含量可用来估计酶切位点的概率例EcoRI,GAATTC:$P=(\frac{g}{2})^2\times(\frac{1-g}{2})^4$, g为GC含量
- Adenylation之后,在adapter ligation阶段连接的是不完整的接头,通常只包含sequencing primer(测序引物结合位点)。
- PCR阶段的引物是“长引物”,其结构从3‘端到5’端依次为:与seq primer互补的序列、barcode(区分样本)、Adapter(flow cell binding序列)
- 采用双索引 dual indexing(i5 和 i7 同时使用)而非单索引,主要基于以下排他性证据的考量:
- 抑制标签漂移 (Index Hopping):在模式芯片(Patterned Flow Cell)上,由于 ExAmp 反应,容易出现 Index 错误延伸。如果只用单索引,发生漂移的 Read 会被分错样本;使用双索引时,只有 i5 和 i7 同时与预设组合匹配的 Read 才会保留。如果出现 A-i5 与 B-i7 这种“杂交”组合,软件会自动将其识别为噪声并剔除。
- 提升样本区分效率:通过组合 8 种 i5 和 12 种 i7,仅需 20 种引物即可区分 8×12=96 个样本,降低了试剂合成成本。
文库质检
质检过程主要围绕三个核心维度:纯度(Purity)、片段大小(Size Distribution)及有效浓度(Functional Concentration)。
1. 浓度与纯度初步评估 (Initial Quantity & Purity)
在文库构建完成后,首先需要确认样本中双链 DNA(dsDNA)的物理总量。
- Qubit (荧光定量法):
- 原理:使用特异性结合 dsDNA 的荧光染料。
- 诊断意义:相比于 Nanodrop(吸光光度法),Qubit 能排除单核苷酸、RNA 及变性 DNA 的干扰,提供更准确的物理浓度。
- Nanodrop (吸光比值法):
- 指标:A260/A280 和 A260/A230。
- 诊断意义:用于检测是否存在有机溶剂(如苯酚、硫氰酸胍)或蛋白质残留,这些物质会抑制后续的集群生成(Cluster Generation)
2. 片段大小分布检测 (Size Distribution)
这是识别文库构建是否符合预期(如酶切是否完全、接头连接是否成功)的核心步骤。
-
常用工具:Agilent 2100 Bioanalyzer, TapeStation 或 Fragment Analyzer。
-
观察指标:
- 主峰位置 (Main Peak):应位于“插入片段长度 + 接头长度”的理论值。
- 接头二聚体 (Adapter Dimer):在 ~120-140 bp 处是否存在窄峰。
- 引物残留 (Primer Residue):在 <80 bp 处是否存在异常。
| 图形特征 | 可能原因 |
|---|---|
| 120bp有高尖峰 | 接头过量或磁珠筛选不彻底。 |
| 高分子量区域有连续拖尾 | PCR循环数过多导致过度扩增,产生异源双链(Heteroduplex)。 |
| 峰形弥散且偏小 | DNA发生降解,或酶切时间过长导致过度消化。 |
3. 有效浓度定量 (qPCR - Functional Quantitation)
这是文库上机前的金标准。
-
核心逻辑:Qubit 测得的是物理浓度(所有 dsDNA),而 qPCR 测得的是有效浓度。
-
引物设计:qPCR 引物直接针对 P5 和 P7(Flow Cell 结合位点)。
-
诊断价值:
- 只有两端都成功连接了接头的分子才会被 qPCR 扩增。
- 计算比值 ($R=C_{qPCR}/C_{Qubit}$):如果 R 显著偏低,说明大部分 DNA 片段未成功连接接头,或者接头连接后发生了严重损伤。
- 测序公司根据 qPCR 的结果计算摩尔浓度,进行文库混合(Pooling)。
4. 文库混样与最终上机 (Pooling & Loading)
在确认单个文库合格后,公司会根据索引(Index/Barcode)平衡各样本比例:
- 摩尔等量混合:确保每个样本在 Flow Cell 上占用的测序通道比例一致。
- 上机浓度标准化:稀释至最终上机浓度(如 1.8 pM 或更低,取决于测序平台)。
混样(Pooling):实现多样本并行测序
混样的目标是将多个带不同 Barcode(Index)的文库按比例混合到同一个离心管中,以便在同一条测序通道(Lane)中同时运行。
测序下机后的数据量(reads数)直接取决于上机时该样本分子的摩尔比例。因此必须将质量浓度C(ng/µl)转换为摩尔浓度M(nM)。
\[M=\frac{C\times10^6}{L\times660}\]L为Bioanalyzer测得的文库平均片段长度(bp)。660为双链DNA的平均分子量(g/mol/bp)。
操作步骤
- 稀释标准值:将所有待混样的文库分别稀释到统一的摩尔浓度(例如 4 nM 或 10 nM)。
- 物理混合:取相同体积的已稀释文库混合。
- 二次质检:对混合后的文库(Pool)再次进行 Qubit 和 qPCR 定量,确保混合液的总体有效浓度符合上机要求。
上机(Loading):从试管到芯片
上机是指将混合好的文库加载到测序仪的载体(Flow Cell)上的过程。
操作步骤
- 变性(Denaturation)
- 物理状态变化:使用 NaOH 将双链文库解链为单链 DNA (ssDNA)。
- 目的:只有单链分子才能与 Flow Cell 表面固定的引物进行碱基互补配对。
- 杂交(Hybridization)
- 原理:Flow Cell 表面植被了海量的 P5 和 P7 寡核苷酸序列。变性后的文库末端(P5/P7 互补端)会通过氢键结合在芯片表面。
- 集群生成(Cluster Generation)
- 过程:通过桥式 PCR(Bridge Amplification)或排斥扩增(Exclusion Amplification, ExAmp),将每一个结合在芯片上的单分子原位扩增成一个包含数千个相同分子的“簇(Cluster)”。
- 意义:测序仪的光学传感器无法捕捉单分子发出的微弱荧光信号,必须通过扩增形成信号集群。