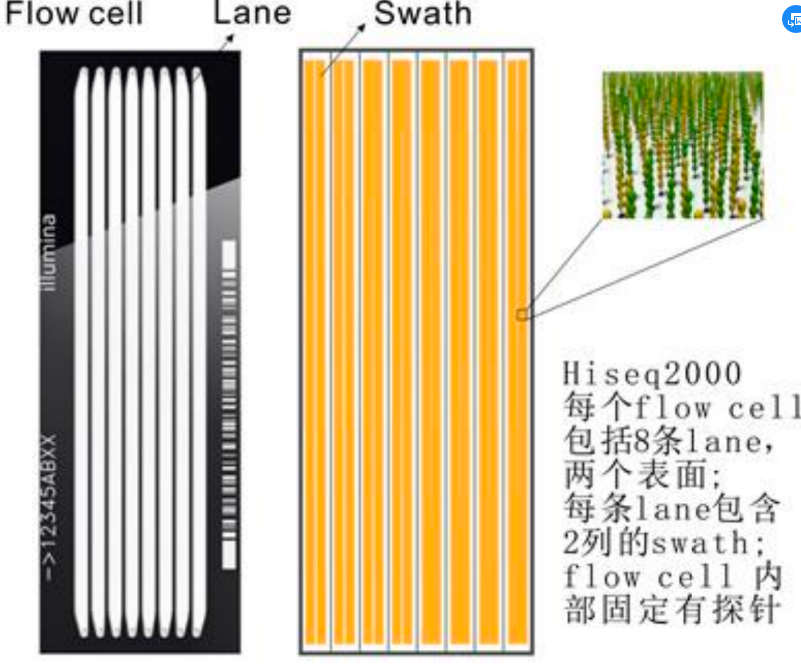
记录了二代测序的原理。
常见测序方法
- Illumina SBS:边合成边测序技术,桥式PCR扩增文库,读长通常在100-300bp之间。
-
- 优势: 成本低、通量高、准确率高(>99.9%)。
-
- 应用: 适用于检测SNP(单核苷酸多态性)、小片段Indel(插入/缺失)、CNV(拷贝数变异)等。
- 代表仪器: Illumina NovaSeq, HiSeq, NextSeq系列。
- 第三代长度长测序
- PacBio SMRT:单分子实时测序,
- Oxford Nanopore Technologies (ONT):纳米孔测序,直接读取单分子DNA,通过纳米孔检测电流变化。
- 优势: 读长长(可达kb至Mb级别),适合解决复杂区域(如高重复序列、SV结构性变异、单倍型分析)。
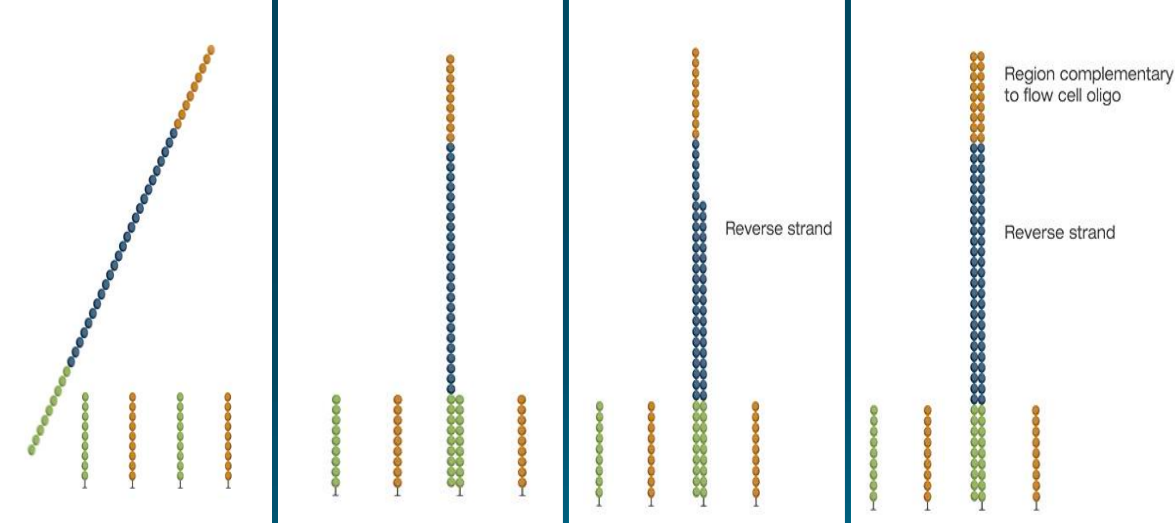
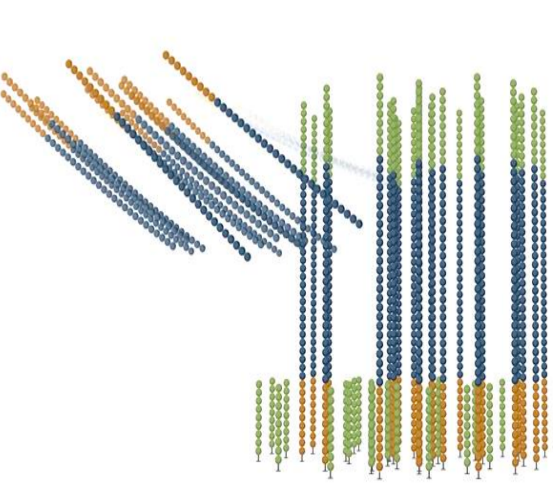
桥式扩增
- 待测DNA片段固定在流动槽的探针上。
- 开启桥式扩增第一个循环
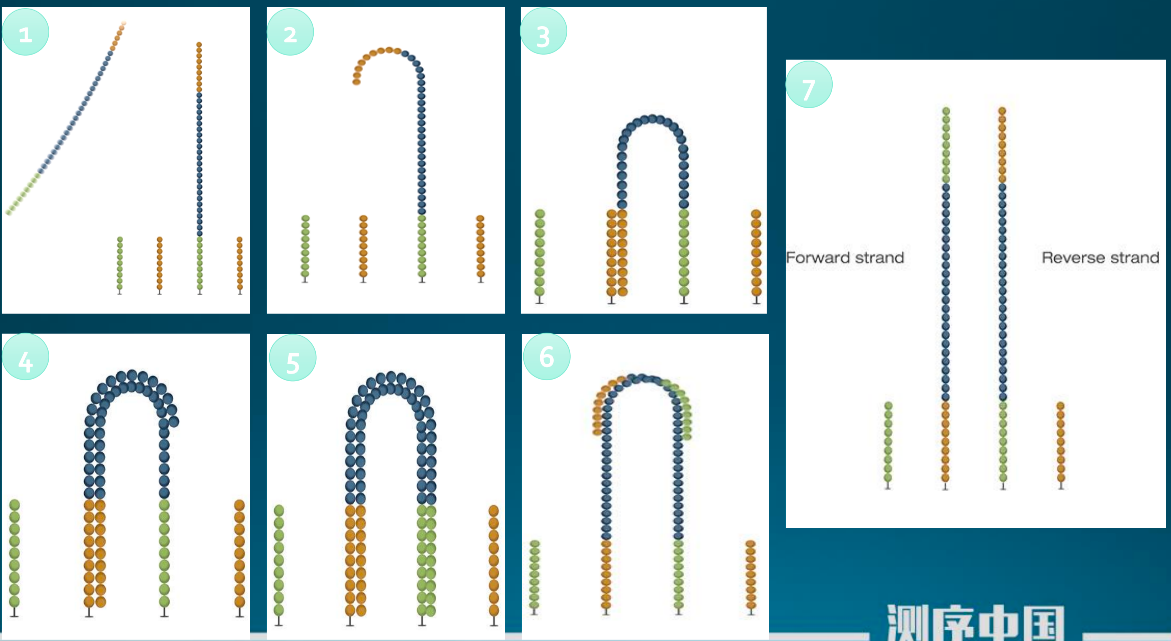
- 扩增至N个循环
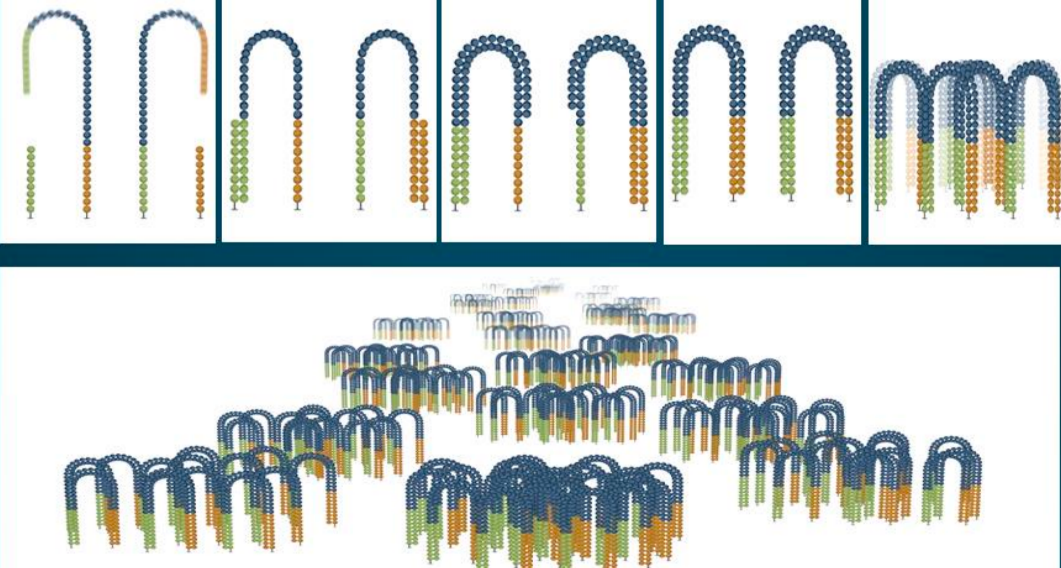
- 仅保留forward/reverse链。
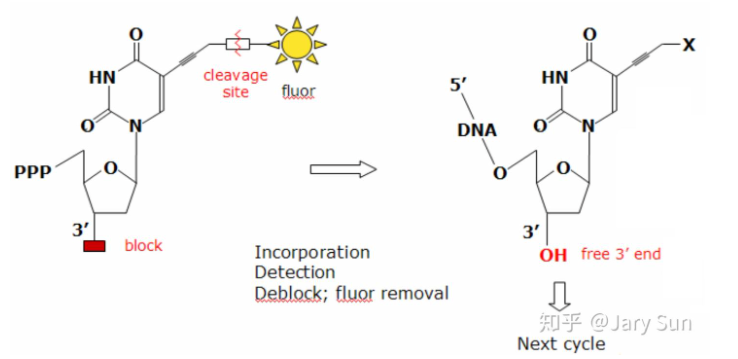
- 边合成边测序:使用的是带有可逆阻断基团的 dNTP(不同于 ddNTP)。其 3’ 端被一个可以化学切除的基团保护。 这种阻断是暂时且可逆的。每轮循环加入一个碱基并拍照后,通过化学试剂切除阻断基团,使 3’ 端恢复羟基,从而继续下一轮合成。
乳液PCR
Roche454,SOLiD,Ion Torrent都是这类。 DNA片段与dNTP、引物和聚合酶包在一个油滴中,独立进行PCR扩增。 优势:
- 每个测序反应都在PTP板上独立的小孔中进行,能大大降低相互间的干扰和测序偏差
- 测序读长,平均读长可达400bp,最长可达800bp 劣势:
- 无法准确测量同聚物长度,如当序列中存在类似于PolyA的情况时,测序反应会一次加入多个T,而所加入的T的个数只能通过荧光强度推测获得,可能导致结果不准
- 测序成本相对较高
ABI Solid 边连接边测序
连接酶测序
- 8碱基单链荧光探针混合物与单链DNA模板链配对完成,发出代表第1,2位碱基的荧光信号,然后在5和第6位碱基之间进行切割,移除荧光信号,即第一轮测序获得第1、2位,第二次是第6、7位……在测到末尾,将新合成的链变性洗脱掉
- 接着用引物n-1进行第二轮测序,发出代表第0,1位碱基的荧光信号,然后在5和第6位碱基之间进行切割,移除荧光信号,即第二轮测序获得第0、1位,第二次是第5、6位……在测到末尾,将新合成的链变性洗脱掉
- 依此类推,用n-2,n-3,n-4继续测序(接入长度为5bp的特殊探针bridge probe),五轮测序后即完成所有位置的碱基测序,且每个位置的碱基均被检测了两次 优势:
- 每个碱基为双次检测,原始测序准确性高达99.94%,而15x覆盖率的准确性更是达到了99.999% 劣势:
- 读长短(2×50bp),后续序列拼接比较复杂
- 鉴于其是双碱基确定一个荧光信号,荧光解码阶段,一旦发生错误就容易产生连锁的解码错误
半导体测序
ION芯片测序
- 将处理好的磁珠固定在高密度半导体芯片的小孔中,与Roche454技术类似,一个小孔即为一个反应池
- 依次加入4种dNTP,配对成功后会释放出一个氢离子,反应池中PH发生改变
- 微电极检测PH变化,检测到的H+离子信号转化为数字信号,从而读出DNA序列
- 洗脱后进入下一轮碱基测序
优势:
- 将化学信号转换为数字信号,不需要昂贵的物理成像等设备,成本低、操作简单、速度快
- 同聚物准确测量得到改善,DNA链上有若干相同碱基时,会检测到电压加倍,通过优化酶聚合酶,如新推出的Hi-Q酶聚合反应非常快,产生的PH值变化的峰更高、更尖、更利于判读,提高判读Homoploymer区域的准确性 劣势:
- 通量较低,仅适合用于小基因组和外显子验证等测序。
华大DNB
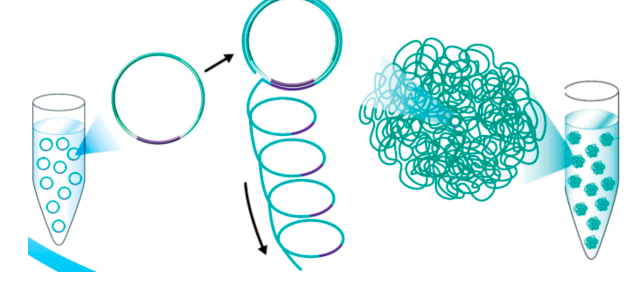 DNA片段扩增(纳米球DNB技术)
1. DNA片段两段加接头
2. 变性处理后分离得到单链DNA
3. 加入与接头序列互补的连接引物,使单链DNA成环,连接酶连接两个接头
4. 在聚合酶的作用下沿着引物进行连续不断的滚环扩增
5. 上百份拷贝都在一股新DNA上,形成一团DNA纳米球(DNB, DNA Nano Ball)
DNA纳米球附着芯片
DNB纳米球经过装载技术固定在阵列化(Patterned Array)的硅芯片上,芯片每个位点的蛋白质自动附着上去一个DNB纳米球
测序
1. DNA分子锚和荧光探针在DNB上进行聚合
2. 高分辨率成像系统对光信号进行采集
3. 光信号经过数字化处理后即可获得待测序列
优势:
DNA片段扩增(纳米球DNB技术)
1. DNA片段两段加接头
2. 变性处理后分离得到单链DNA
3. 加入与接头序列互补的连接引物,使单链DNA成环,连接酶连接两个接头
4. 在聚合酶的作用下沿着引物进行连续不断的滚环扩增
5. 上百份拷贝都在一股新DNA上,形成一团DNA纳米球(DNB, DNA Nano Ball)
DNA纳米球附着芯片
DNB纳米球经过装载技术固定在阵列化(Patterned Array)的硅芯片上,芯片每个位点的蛋白质自动附着上去一个DNB纳米球
测序
1. DNA分子锚和荧光探针在DNB上进行聚合
2. 高分辨率成像系统对光信号进行采集
3. 光信号经过数字化处理后即可获得待测序列
优势:
- DNB通过增加待测DNA的拷贝数而增强了信号强度,从而提高测序准确度
- 不同于PCR的指数扩增,滚环扩增技术的扩增错误不会累积
- DNB与芯片上活化位点的大小相同,每个位点只固定一个DNB,保证信号点之间不产生相互干扰
- 阵列化测序芯片和DNB测序技术的结合,使得成像系统像素和测序芯片的面积得到最大化利用
- 核心技术:DNB技术、Patterned array技术、cPAS技术
二代测序出现错误的原因
1. 信号积累与信噪比退化(生化层面)
Phasing 和 Pre-phasing 属于这一类。其本质是分子群落(Cluster)的非同步化。
-
Phasing (滞后):化学反应不完全(末端修饰未脱落或未延伸),导致部分分子留在 n−1 步。
-
Pre-phasing (超前):末端阻断失效,导致部分分子直接跳到 n+1 步。
-
后果:随着循环数(Cycle)增加,噪声呈指数级积累。这就是为什么 Illumina 读长通常限制在 150-300bp 的物理瓶颈——到后面,“杂音”盖过了“主音”,Q30 质量值断崖式下跌。
个人理解:如果一个集群(Cluster)中有 10% 的分子发生了 Phasing(滞后),那么在第 10 轮成像时,这 10% 的分子发出的其实是第 9 轮的信号($I_{n−1}$)。测序软件(Basecaller)每一轮都会强行识别一个碱基。如果$I_{n−1}$的背景噪声太强,导致识别错误,结果就是在该位点记下一个错误的碱基(替换错误),但不会改变总读长。
2. 空间重叠与集群密度(光学层面)
除了信号的时间同步问题,还有空间上的分辨问题。
-
Over-clustering(过饱和):如果上机浓度(Loading concentration)过高,Flow Cell 上的集群会靠得太近。
-
后果:光学镜头无法分辨两个相邻集群的荧光信号,导致坐标定位(Template Generation)失败或颜色串扰。这种错误通常发生在测序的最初几个循环。
-
Under-clustering(密度过低):虽然错误率低,但数据产量极低,导致有效信息不足。
个人理解:
- 非模式芯片(non-patterned flow cell)中,DNA 片段随机结合在探针表面,如果浓度太高,扩增后的两个cluster可能在空间上发生物理重叠导致相机无法区分,信号被丢弃或识别错误。
- 而模式芯片(patterned flow cell),如NovaSeq,通过Exclusion Amplification(ExAmp)“排除扩增”确保一个孔只有一个cluster(一个孔只会被一个DNA片段填满),但浓度过高会导致不同DNA片段同时进入一个孔并开始扩增(co-occupancy),导致信号无法正确识别。
3. 序列偏好性与系统性偏差(序列层面)
某些特定的 DNA 序列天生难以被准确测序,这与 Phasing 无关,而与生化动力学有关。
- GC 含量偏好(GC Bias):
- 低 GC 或高 GC 区域:在集群扩增(Bridge PCR)过程中,这些区域的扩增效率极低,导致信号强度弱。
- 同聚物(Homopolymers):
- 虽然 Illumina 对 AAAAA 这种序列比 454 或 Ion Torrent 系统更稳定,但在极长同聚物中,依然会因为聚合酶滑移(Polymerase Slippage)导致插入或缺失错误(Indel)。
- G-quadruplex(G-四联体):复杂的二级结构会物理性阻碍聚合酶的延伸。
4. 实验室引入的“人造”错误(样本准备层面)
这些错误在测序前就已经存在于文库中。
-
PCR 嵌合体(Chimeras)与错配:文库构建时的扩增步骤会引入碱基错配。平均而言,高保真酶的错误率约为 10−6,虽然低,但在深度测序中会被放大。
-
Index Hopping(标签漂移):在 Patterned Flow Cell 上,游离的接头可能导致 A 样本的 Read 被错误地标记为 B 样本。 个人理解:文库构建过程中,接头(Adapter)是过量加入的。即使经过两轮磁珠纯化仍然会有极微量的游离接头或引物残留。上机后,flow cell存在已结合的单链,游离接头,游离样本,以及高活性的DNA聚合酶。游离的接头B以文库A为模板通过其互补的P5/P7延伸,最终导致本属于样本A的片段接上了B的barcode。
光信号转换为数字信号
测序仪记录的是图像光信号(tiff) 需要转换为cif location,再转为BCL(进行stats control filter),最后得到fastq
BCL:BaseCalL,二进制文件,包含了每个circles和tite的碱基信息和质量分数,是真正意义上的RawData,由仪器的实时分析软件(RTA)生成
目前主流的数据拆分软件是bcl2fastq和bcl-convert
Bcl2fastq为旧版本的数据拆分软件,拆分速度较慢,目前已不再更新
Bcl-convert具有算法上的优化,拆分速度快,资源消耗小
不同软件拆分出的fastq ID不同